Potrebbero piacerti anche
- Lab ManualDocumento4 pagineLab ManualHasam Tanveer H T MNessuna valutazione finora
- USP - Choline ChlorideDocumento3 pagineUSP - Choline Chloridehh.8968Nessuna valutazione finora
- Titration Part 1Documento5 pagineTitration Part 1takomolyentinNessuna valutazione finora
- Experiment 1: CARBOHYDRATES: A. Qualitative Tests and Analysis of UnknownDocumento21 pagineExperiment 1: CARBOHYDRATES: A. Qualitative Tests and Analysis of UnknownJamie FloresNessuna valutazione finora
- AOAC970Documento2 pagineAOAC970Luisa Fernanda GonzálezNessuna valutazione finora
- 09.11.2010 MethodologyDocumento16 pagine09.11.2010 MethodologyavvaimsvijayaNessuna valutazione finora
- No 3Documento12 pagineNo 3Punit Ratna ShakyaNessuna valutazione finora
- Experiment 10: H TH H THDocumento8 pagineExperiment 10: H TH H THPakistan ideologueNessuna valutazione finora
- Name Shatanshu Raj Student ID: SE21UCSE199 Batch Number: A4Documento8 pagineName Shatanshu Raj Student ID: SE21UCSE199 Batch Number: A4Kadarla ShashankNessuna valutazione finora
- Pharmaceutical CGMP Guidelines Water TestingDocumento24 paginePharmaceutical CGMP Guidelines Water TestingloisetapiceriaNessuna valutazione finora
- Self Directed Learning Sch3u Lab ManualDocumento20 pagineSelf Directed Learning Sch3u Lab Manualapi-281434216Nessuna valutazione finora
- Problem Set 1Documento3 pagineProblem Set 1Lu JunqueiraNessuna valutazione finora
- Análisis de SuelosDocumento10 pagineAnálisis de SuelosCristian CarrascoNessuna valutazione finora
- Quantitative Estimation of Amino Acids by Ninhydrin: TheoryDocumento11 pagineQuantitative Estimation of Amino Acids by Ninhydrin: TheoryNamrata KulkarniNessuna valutazione finora
- Total Dissolved Solids ProcedureDocumento13 pagineTotal Dissolved Solids Procedurehemavathi jayNessuna valutazione finora
- Standard Analytical ProceduresDocumento80 pagineStandard Analytical Proceduresengr_afsoomro3147Nessuna valutazione finora
- Antacid Suspension With Oxetacaine & SimethiconeDocumento5 pagineAntacid Suspension With Oxetacaine & SimethiconePatricia Joyce Malabanan Sunglao100% (1)
- Hexanes: Chemical Names Chemical Formula Formula WeightDocumento6 pagineHexanes: Chemical Names Chemical Formula Formula WeightRustika SafitriNessuna valutazione finora
- Chemistry Practicals First YearsDocumento65 pagineChemistry Practicals First Yearskokimesh0% (1)
- CodDocumento3 pagineCodjaineemNessuna valutazione finora
- GB5009. 12 2010 Determination of Lead in FoodsDocumento18 pagineGB5009. 12 2010 Determination of Lead in FoodsIvone SulistyaNessuna valutazione finora
- Lab 1 AcidityDocumento8 pagineLab 1 AcidityEngr Arafat QubatiNessuna valutazione finora
- New LowryDocumento6 pagineNew LowrymourighoshNessuna valutazione finora
- Experiment No. 3 Volumetric TransferDocumento14 pagineExperiment No. 3 Volumetric TransferJoemar SubongNessuna valutazione finora
- Total AlkalinityDocumento7 pagineTotal Alkalinityfakher adnanNessuna valutazione finora
- General Chemistry: Lab 4: Thermodynamics IIDocumento6 pagineGeneral Chemistry: Lab 4: Thermodynamics IIAsif ShahNessuna valutazione finora
- E13 Volumetric Analysis Acid / Base Titrations: The TaskDocumento7 pagineE13 Volumetric Analysis Acid / Base Titrations: The TaskbokjooooNessuna valutazione finora
- Alkaloid, Tanin, Flavonoid and Saponin AnalysisDocumento7 pagineAlkaloid, Tanin, Flavonoid and Saponin AnalysisNiken Ayu PermatasariNessuna valutazione finora
- PagesfromAnalytical Techniques in Biochemist-17Documento3 paginePagesfromAnalytical Techniques in Biochemist-17Alex WasabiNessuna valutazione finora
- Anachm 2 - Expt 1Documento5 pagineAnachm 2 - Expt 1CNessuna valutazione finora
- Titration - Lab-ManualDocumento9 pagineTitration - Lab-ManualVN BomXanhNessuna valutazione finora
- Experiment 10 Titration v2Documento14 pagineExperiment 10 Titration v2Renu ReenuNessuna valutazione finora
- Fluoride Electrode ManualDocumento26 pagineFluoride Electrode ManualNilesh SomtiyaNessuna valutazione finora
- Enzyme-Technology-and-Biokinetics-Lab-Manual-BT-47L For Food LabDocumento21 pagineEnzyme-Technology-and-Biokinetics-Lab-Manual-BT-47L For Food Labmasre semagnNessuna valutazione finora
- Experiment 1&2Documento8 pagineExperiment 1&2Fatima AhmedNessuna valutazione finora
- Bioseparation Lab ManualDocumento21 pagineBioseparation Lab ManualJaganathan MkNessuna valutazione finora
- Assay of Ractopamine HCL 2Documento7 pagineAssay of Ractopamine HCL 2Enpex LaboratoriesNessuna valutazione finora
- Acid Base TitrationDocumento12 pagineAcid Base Titrationdonna benitoNessuna valutazione finora
- Expt-4 PotentiometryDocumento3 pagineExpt-4 PotentiometryAhyessa CastilloNessuna valutazione finora
- Lab Manual PDFDocumento84 pagineLab Manual PDFSohail AhamdNessuna valutazione finora
- Dioctyl SulfateDocumento3 pagineDioctyl SulfateZach CaesarNessuna valutazione finora
- 2 5 23 Sialic Acid in Polysaccharide VaccinesDocumento1 pagina2 5 23 Sialic Acid in Polysaccharide VaccinesLTV TM DVNessuna valutazione finora
- Exp. 1Documento7 pagineExp. 1علي عقيل مهديNessuna valutazione finora
- Exp 2 Analysis of Unknown Acetic Acid Solution As245 Applied ChemistryDocumento10 pagineExp 2 Analysis of Unknown Acetic Acid Solution As245 Applied ChemistryNaz HelmiNessuna valutazione finora
- Lab Report ExampleDocumento7 pagineLab Report Examplealiswheeler12Nessuna valutazione finora
- Experiment 2Documento5 pagineExperiment 2aeydrusNessuna valutazione finora
- AP LACTOSE MonohydrateDocumento4 pagineAP LACTOSE MonohydrateAde YuLianiNessuna valutazione finora
- Experiment 2 Analysis of An Unknown Vinegar Sample ObjectivesDocumento5 pagineExperiment 2 Analysis of An Unknown Vinegar Sample ObjectivesVirginia DukeNessuna valutazione finora
- Experiment Name: Analysis of Non-Alcoholic Beverages. Sample: Lime SquashDocumento10 pagineExperiment Name: Analysis of Non-Alcoholic Beverages. Sample: Lime SquashJoyita khanNessuna valutazione finora
- Acid Base TitrationsDocumento6 pagineAcid Base Titrationssadya98100% (1)
- Laboratory Experiment #1common Laboratory Operations (Part 2)Documento11 pagineLaboratory Experiment #1common Laboratory Operations (Part 2)Monica RilveriaNessuna valutazione finora
- Ascorbic Acid Titration Summer 2019 One PeriodDocumento9 pagineAscorbic Acid Titration Summer 2019 One PeriodTaiga KagamiNessuna valutazione finora
- Lycopene Extract From TomatoDocumento7 pagineLycopene Extract From TomatoShayne PalalayNessuna valutazione finora
- Physical Pharmacy Lab (PHA205L)Documento15 paginePhysical Pharmacy Lab (PHA205L)moin4cuetNessuna valutazione finora
- Advanced Pharmaceutical analysisDa EverandAdvanced Pharmaceutical analysisValutazione: 4.5 su 5 stelle4.5/5 (2)
- Practical Manual of Analytical ChemistryDa EverandPractical Manual of Analytical ChemistryValutazione: 4.5 su 5 stelle4.5/5 (3)
- Standard methods for the examination of water and sewageDa EverandStandard methods for the examination of water and sewageNessuna valutazione finora
- Practical Handbook of Pharmaceutical Chemistry for M.PharmDa EverandPractical Handbook of Pharmaceutical Chemistry for M.PharmNessuna valutazione finora
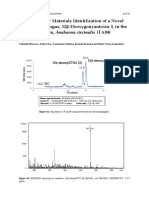
- Supplementary Materials Identification of a Novel Saxitoxin Analogue, 12β-Deoxygonyautoxin 3, in the Cyanobacterium, Anabaena circinalis (TA04)Documento4 pagineSupplementary Materials Identification of a Novel Saxitoxin Analogue, 12β-Deoxygonyautoxin 3, in the Cyanobacterium, Anabaena circinalis (TA04)Mheira VillahermosaNessuna valutazione finora
- Philippine Priority Chemicals List PCL PDFDocumento7 paginePhilippine Priority Chemicals List PCL PDFKaren Feyt MallariNessuna valutazione finora
- Dao 2005 27Documento3 pagineDao 2005 27Jannine TrinidadNessuna valutazione finora
- Solid-State NMR Studies of The Interactions Between Cationic MembDocumento179 pagineSolid-State NMR Studies of The Interactions Between Cationic MembMheira VillahermosaNessuna valutazione finora
- Enzyme InhibitionDocumento13 pagineEnzyme InhibitionMheira Villahermosa100% (1)
- Encapsulation of Gases in Solid StateDocumento2 pagineEncapsulation of Gases in Solid StateMheira VillahermosaNessuna valutazione finora
- Problemset1a AnswersDocumento4 pagineProblemset1a AnswersMheira VillahermosaNessuna valutazione finora
- The BEST SOURCE - Spectroscopic - TechniquesDocumento27 pagineThe BEST SOURCE - Spectroscopic - TechniquesMheira VillahermosaNessuna valutazione finora
- Enzyme KineticsDocumento4 pagineEnzyme KineticsMheira VillahermosaNessuna valutazione finora
- Take Home Exam 1 (2013)Documento1 paginaTake Home Exam 1 (2013)Mheira VillahermosaNessuna valutazione finora
- 2 - UV-VIS - Take Home Exam, Num5Documento6 pagine2 - UV-VIS - Take Home Exam, Num5Mheira VillahermosaNessuna valutazione finora
- ElectrogravimetryDocumento5 pagineElectrogravimetryMheira VillahermosaNessuna valutazione finora
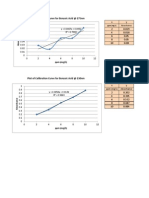
- CHEM127.1 - Exp4 (Calib Curve - Benzoic Acid)Documento5 pagineCHEM127.1 - Exp4 (Calib Curve - Benzoic Acid)Mheira VillahermosaNessuna valutazione finora
- Plot of Calibration Curve For Caffeine at 275nm: PPM (MG/L)Documento1 paginaPlot of Calibration Curve For Caffeine at 275nm: PPM (MG/L)Mheira VillahermosaNessuna valutazione finora
- Voltammetry and PolarographyDocumento21 pagineVoltammetry and PolarographyMheira Villahermosa100% (2)
- Chem127.1 - Exp3 (Quali Uv-Vis Data)Documento4 pagineChem127.1 - Exp3 (Quali Uv-Vis Data)Mheira VillahermosaNessuna valutazione finora
- FRET PresentationDocumento25 pagineFRET PresentationMheira VillahermosaNessuna valutazione finora
- TRM Reb670Documento490 pagineTRM Reb670jayapalNessuna valutazione finora
- Fdot Modifications To LRFD Specifications For Structural Supports For Highway Signs, Luminaires and Traffic Signals (Lrfdlts-1)Documento26 pagineFdot Modifications To LRFD Specifications For Structural Supports For Highway Signs, Luminaires and Traffic Signals (Lrfdlts-1)kayshephNessuna valutazione finora
- L28-32H GenSet TierII PDFDocumento824 pagineL28-32H GenSet TierII PDFHelder Pinto100% (1)
- Chloride LINEAR MK II - Service ManualDocumento32 pagineChloride LINEAR MK II - Service Manualfabio.perazzoloNessuna valutazione finora
- Markov ChainDocumento16 pagineMarkov Chainnaveenk903Nessuna valutazione finora
- 2.lecture 1-Basics and PrecedenceDocumento30 pagine2.lecture 1-Basics and PrecedenceBhavesh ReddyNessuna valutazione finora
- Distance Determination For An Automobile Environment Using Inverse Perspective Mapping in OpenCVDocumento6 pagineDistance Determination For An Automobile Environment Using Inverse Perspective Mapping in OpenCVCristian StrebaNessuna valutazione finora
- Beckman Coulter GenomeLab TroubleshootDocumento56 pagineBeckman Coulter GenomeLab TroubleshootChrisNessuna valutazione finora
- Latex TutorialDocumento27 pagineLatex TutorialSalman AhmadNessuna valutazione finora
- Heat and Thermodynamics According To KPK Textbook & Sindh TextbookDocumento15 pagineHeat and Thermodynamics According To KPK Textbook & Sindh Textbookswatmiandam44Nessuna valutazione finora
- Ijtmsr201919 PDFDocumento5 pagineIjtmsr201919 PDFPrakash InturiNessuna valutazione finora
- CHE504 - Lab Report On Gas Absorption L8 PDFDocumento23 pagineCHE504 - Lab Report On Gas Absorption L8 PDFRakesh KumarNessuna valutazione finora
- Tech Tip: How To Test For An Alternator Voltage DropDocumento2 pagineTech Tip: How To Test For An Alternator Voltage DropainginerNessuna valutazione finora
- StructSteel Takeoff InstructionsDocumento56 pagineStructSteel Takeoff InstructionsAmrish TyagiNessuna valutazione finora
- Notes: Edited by William AdkinsDocumento6 pagineNotes: Edited by William Adkinsjorge mario durango petroNessuna valutazione finora
- Unit No 1Documento64 pagineUnit No 1Aadil VahoraNessuna valutazione finora
- Bahir Dar Institute of Technology FacultDocumento140 pagineBahir Dar Institute of Technology Facultyared sitotaw100% (1)
- DDP400 Open-Frame and U-Chassis :: ROAL Living EnergyDocumento12 pagineDDP400 Open-Frame and U-Chassis :: ROAL Living EnergyroalscribdNessuna valutazione finora
- Calculations of The EFG Tensor in DTN Using GIPAW With CASTEP and QE SoftwareDocumento12 pagineCalculations of The EFG Tensor in DTN Using GIPAW With CASTEP and QE SoftwareAllen MNessuna valutazione finora
- Properties of Gray Cast Iron - Engineer's HandbookDocumento2 pagineProperties of Gray Cast Iron - Engineer's Handbookkus satria dNessuna valutazione finora
- Chapter Seven: GS Ohlins Installation, Adjustment Tips & TricksDocumento29 pagineChapter Seven: GS Ohlins Installation, Adjustment Tips & TricksWhattonNessuna valutazione finora
- Lesson Plan in Remainders TheoremDocumento5 pagineLesson Plan in Remainders TheoremJune SabatinNessuna valutazione finora
- ReportDocumento1 paginaReportDrew DacanayNessuna valutazione finora
- Digital Image ProcessingDocumento156 pagineDigital Image ProcessingAnushka BajpaiNessuna valutazione finora
- Turbo-Pump Supply System For Liquid-Propellant Rocket EngineDocumento8 pagineTurbo-Pump Supply System For Liquid-Propellant Rocket EngineĐinh Quốc TríNessuna valutazione finora
- Glut 3Documento68 pagineGlut 3Lê Quốc HoàngNessuna valutazione finora
- 235practice Exam 2 AnswerDocumento9 pagine235practice Exam 2 Answernbobs7Nessuna valutazione finora
- Design Constraint ReportDocumento11 pagineDesign Constraint ReportCam MillerNessuna valutazione finora
- Quality For WeldsDocumento9 pagineQuality For WeldsArturs StangainisNessuna valutazione finora
- Product Data Sheet: Control Unit Micrologic 5.0 A, For Masterpact NT/ NW, LSI ProtectionsDocumento3 pagineProduct Data Sheet: Control Unit Micrologic 5.0 A, For Masterpact NT/ NW, LSI ProtectionsEvandro PavesiNessuna valutazione finora