Potrebbero piacerti anche
- Essential Pharmacokinetics: A Primer for Pharmaceutical ScientistsDa EverandEssential Pharmacokinetics: A Primer for Pharmaceutical ScientistsValutazione: 5 su 5 stelle5/5 (1)
- ANS Pharmacology-Cholinergic AgentsDocumento55 pagineANS Pharmacology-Cholinergic AgentsMarc Imhotep Cray, M.D.Nessuna valutazione finora
- Pharmacology Concept BookDocumento357 paginePharmacology Concept Bookram dheerNessuna valutazione finora
- Pharmacology Study NotesDocumento121 paginePharmacology Study Notesninja-2001100% (1)
- Pharmacology MnemonicsDocumento19 paginePharmacology MnemonicsAl-nazer Azer Al100% (5)
- Basic Principles General PharmacologyDocumento47 pagineBasic Principles General Pharmacologysapiah raman100% (2)
- Pharmacology List of DrugsDocumento66 paginePharmacology List of DrugsSohail Adnan100% (2)
- Question Bank For Pharmacology IiDocumento9 pagineQuestion Bank For Pharmacology Iithomasarun2009100% (24)
- Section 5 The Nervous SystemDocumento33 pagineSection 5 The Nervous SystemNasir Javed96% (23)
- Questions - Answer Bassam QQ 1Documento14 pagineQuestions - Answer Bassam QQ 1Bassam SaifNessuna valutazione finora
- PharmacologyDocumento318 paginePharmacologyAamir Sirohi94% (16)
- Pharmacology: Core Curriculum in NephrologyDocumento11 paginePharmacology: Core Curriculum in NephrologyYuppie RajNessuna valutazione finora
- Pharmacology - Lecture Notes, Study Material and Important Questions, AnswersDocumento52 paginePharmacology - Lecture Notes, Study Material and Important Questions, AnswersM.V. TV100% (1)
- Pharmacology NotesDocumento87 paginePharmacology NotesAnonymous anXPzpy4100% (1)
- PharmacologyDocumento19 paginePharmacologyBhanu K Prakash100% (1)
- Respiratory System - Notes (2023!03!15)Documento12 pagineRespiratory System - Notes (2023!03!15)JOSEPH VINCENT100% (1)
- Cholinergic DrugsDocumento32 pagineCholinergic DrugsApt FianNessuna valutazione finora
- General Pharmacology NotesDocumento67 pagineGeneral Pharmacology NotesZehra KhanNessuna valutazione finora
- CalculationDocumento24 pagineCalculationhablet1100% (1)
- Endocrine PharmacologyDocumento42 pagineEndocrine PharmacologyAhmed El SharkawyNessuna valutazione finora
- General Pharmacology (1-7)Documento7 pagineGeneral Pharmacology (1-7)LotfyAdel100% (1)
- Gpat 2020 Achiver 2400 Mcqs Exp - CDocumento376 pagineGpat 2020 Achiver 2400 Mcqs Exp - CRohit patelNessuna valutazione finora
- General Pharmacology - Sources of Drugs and Routes of AdministrationDocumento48 pagineGeneral Pharmacology - Sources of Drugs and Routes of AdministrationDhriti Brahma78% (9)
- Pharma 1.2 - Pharmacokinetics (BHND) PDFDocumento13 paginePharma 1.2 - Pharmacokinetics (BHND) PDFVon Javier Gamatero100% (1)
- Pharmacology Lab ManualDocumento73 paginePharmacology Lab ManualRonald Darwin58% (12)
- Pharmacology Study Notes - Adrenergic DrugsDocumento2 paginePharmacology Study Notes - Adrenergic Drugsstuckaluck83% (6)
- Pharmacology Summary Notes Unit 9Documento9 paginePharmacology Summary Notes Unit 9Denny HempataweeNessuna valutazione finora
- Drug Interaction Can Be Defined As TheDocumento34 pagineDrug Interaction Can Be Defined As TheIstianah Es100% (1)
- Ain Shams University - Pharmacology MCQ Ain Shams (2019 - 2020)Documento144 pagineAin Shams University - Pharmacology MCQ Ain Shams (2019 - 2020)Cristian C BecerraNessuna valutazione finora
- Semester Iv Pharmacology I (BP404 TP) Multiple Choice Questions Chapter 1 & 2Documento34 pagineSemester Iv Pharmacology I (BP404 TP) Multiple Choice Questions Chapter 1 & 2Aman Gurjar100% (1)
- Pharmacology SummaryDocumento32 paginePharmacology Summaryminikatiting95% (22)
- 1 - Pharmaceutical Care Practice - An OverviewDocumento76 pagine1 - Pharmaceutical Care Practice - An OverviewekramNessuna valutazione finora
- ZSMU, Ukraine Pharmacology MCQs by Gankidi Raghavender Reddy,,,Used For Preparation of FMGE (Mci Screening Test) TooDocumento117 pagineZSMU, Ukraine Pharmacology MCQs by Gankidi Raghavender Reddy,,,Used For Preparation of FMGE (Mci Screening Test) Toogrreddy836100% (2)
- Pharmacology Mock Exam MCQDocumento8 paginePharmacology Mock Exam MCQanaeshklNessuna valutazione finora
- Comunity Pharmacy Introduction ModDocumento42 pagineComunity Pharmacy Introduction ModPrity girlNessuna valutazione finora
- Clinical Pharmacology, Toxicology and Poisoning: 1. Drug Metabolism and InteractionsDocumento26 pagineClinical Pharmacology, Toxicology and Poisoning: 1. Drug Metabolism and InteractionsNadia Ancharuz100% (1)
- Pharmacology Practice Test IDocumento27 paginePharmacology Practice Test Isidharta_chatterjeeNessuna valutazione finora
- Pharmacology - Git DrugsDocumento123 paginePharmacology - Git DrugsBenjamin Joel Breboneria75% (4)
- Pharmacology Illustrated NotesDocumento148 paginePharmacology Illustrated NotesShikha Khemani89% (9)
- MCQ'S in Pharmacology Wid AnswersDocumento18 pagineMCQ'S in Pharmacology Wid Answersapi-1991639975% (4)
- Final Pharmacology MCQDocumento12 pagineFinal Pharmacology MCQSiraj Choudhari0% (1)
- ++++medicinal ChemistryDocumento229 pagine++++medicinal Chemistrynadjibwassim100% (1)
- Pharmacology Notes PDFDocumento434 paginePharmacology Notes PDFAbdelrahman Mamdouh88% (32)
- Pharmacology Misbah PDFDocumento238 paginePharmacology Misbah PDFRiham Khamis86% (7)
- ANS Physiology and PharmacologyDocumento79 pagineANS Physiology and PharmacologyMarc Imhotep Cray, M.D.100% (1)
- Pharmacology NotesDocumento48 paginePharmacology NotesBheru Lal100% (1)
- Basic PharmacologyDocumento48 pagineBasic PharmacologyEmily Mccarthy100% (2)
- Cholinergic Antagonists "Anticholinergic Drugs" (ParasympatholyticsDocumento32 pagineCholinergic Antagonists "Anticholinergic Drugs" (ParasympatholyticsmiznahNessuna valutazione finora
- Instant Clinical PharmacologyDocumento112 pagineInstant Clinical Pharmacologycelecosib100% (3)
- A Textbook of Clinical Pharmacology and TherapeuticsDocumento476 pagineA Textbook of Clinical Pharmacology and TherapeuticsAli Hassan100% (1)
- Biopharmaceutics and PharmacokineticsDocumento76 pagineBiopharmaceutics and PharmacokineticsEswar Gupta Maddi100% (4)
- Fundamentals of Medicinal Chemistry and Drug MetabolismDa EverandFundamentals of Medicinal Chemistry and Drug MetabolismNessuna valutazione finora
- NAPLEX Practice Question Workbook: 1,000+ Comprehensive Practice Questions (2023 Edition)Da EverandNAPLEX Practice Question Workbook: 1,000+ Comprehensive Practice Questions (2023 Edition)Valutazione: 4.5 su 5 stelle4.5/5 (3)
- Pharmacology: A Companion Handbook with Illustrations: Pharmacology: A Companion Handbook with IllustrationsDa EverandPharmacology: A Companion Handbook with Illustrations: Pharmacology: A Companion Handbook with IllustrationsValutazione: 2 su 5 stelle2/5 (1)
- Senior Pharmacist: Passbooks Study GuideDa EverandSenior Pharmacist: Passbooks Study GuideNessuna valutazione finora
- Naplex Complete Study Outline A Topic-Wise Approach DiabetesDa EverandNaplex Complete Study Outline A Topic-Wise Approach DiabetesValutazione: 4 su 5 stelle4/5 (2)
- Concepts and Prevention of Disease PDFDocumento41 pagineConcepts and Prevention of Disease PDFAna Rika Javier Harder100% (1)
- Sensitivity of Pseudomonas Aeroginosa To Libas (Spondias Pinnata) Leaves ExtractDocumento6 pagineSensitivity of Pseudomonas Aeroginosa To Libas (Spondias Pinnata) Leaves ExtractAna Rika Javier HarderNessuna valutazione finora
- Diuretics TabulationDocumento3 pagineDiuretics TabulationAna Rika Javier HarderNessuna valutazione finora
- Trichosanthes Dioica Roxb (Patal), Cucurbita Maxima (Pumpkin) and Abelmoschus Esculentus Moench (Okra) Plant SeedsDocumento49 pagineTrichosanthes Dioica Roxb (Patal), Cucurbita Maxima (Pumpkin) and Abelmoschus Esculentus Moench (Okra) Plant SeedsAna Rika Javier Harder100% (1)
- Bio 1M03Documento73 pagineBio 1M03brady47Nessuna valutazione finora
- Tutorial Nematoda 1 2019Documento22 pagineTutorial Nematoda 1 2019Andre HimawanNessuna valutazione finora
- Yearbook 2016Documento308 pagineYearbook 2016achintNessuna valutazione finora
- BIO270 Prelab 1 Assignment 2014Documento8 pagineBIO270 Prelab 1 Assignment 2014noahyuyanNessuna valutazione finora
- Reflection On Living Soil - Manaloto - II-24Documento1 paginaReflection On Living Soil - Manaloto - II-24Ren Manaloto0% (1)
- Origin of Eukaryotic Cell PDFDocumento8 pagineOrigin of Eukaryotic Cell PDFaco_golarNessuna valutazione finora
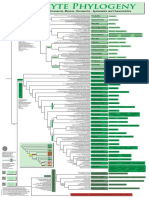
- Filogenia de Briofitos SensulatoDocumento1 paginaFilogenia de Briofitos SensulatoYuyitoS2714Nessuna valutazione finora
- CVS - Charts & DiagramsDocumento20 pagineCVS - Charts & DiagramsMamathaNessuna valutazione finora
- Q4 Week 3 4 GenBio2 EditedDocumento12 pagineQ4 Week 3 4 GenBio2 EditedXyreen GalicinaoNessuna valutazione finora
- Eysenck Personality TheoryDocumento11 pagineEysenck Personality TheoryKarthika RsNessuna valutazione finora
- Bioenergetics and Biological Oxidation 2: Biochemistry: Shift 1 - Trans 4Documento9 pagineBioenergetics and Biological Oxidation 2: Biochemistry: Shift 1 - Trans 4Gemay DanglayNessuna valutazione finora
- Activity Book 2Documento14 pagineActivity Book 2Josune Arévalo75% (4)
- Ethnic Minorities and Mental HealthDocumento33 pagineEthnic Minorities and Mental HealthAdrianoMascalzoneNessuna valutazione finora
- Farmers and Breeders Rights ProjectDocumento15 pagineFarmers and Breeders Rights ProjectJyoti GautamNessuna valutazione finora
- Craetagusarticle PDFDocumento7 pagineCraetagusarticle PDFKatty AcostaNessuna valutazione finora
- In MaizeDocumento9 pagineIn MaizeAnytha Purwareyni UmbasNessuna valutazione finora
- Module 5 Anatomy I Splanchnology 1Documento17 pagineModule 5 Anatomy I Splanchnology 1neannaNessuna valutazione finora
- Nano Dox CarrierDocumento7 pagineNano Dox CarrierMuanfan SuwanNessuna valutazione finora
- Biological Science (BS) : E:/Section-9/Project Proposal/project Proposal 2019-20/secoundary List/BS-2019-2020Documento46 pagineBiological Science (BS) : E:/Section-9/Project Proposal/project Proposal 2019-20/secoundary List/BS-2019-2020Shohel RanaNessuna valutazione finora
- 1.wall of ThoraxDocumento14 pagine1.wall of ThoraxChandru ANessuna valutazione finora
- Menstruation and FertilizationDocumento54 pagineMenstruation and FertilizationJanaica JuanNessuna valutazione finora
- XI NEET - Dakshana Test ScheduleDocumento5 pagineXI NEET - Dakshana Test SchedulePoornima K TNessuna valutazione finora
- Vital SignsDocumento15 pagineVital SignsSelene HmpNessuna valutazione finora
- Nail Structure and Growth: Chapter OutlineDocumento8 pagineNail Structure and Growth: Chapter Outlinebritiena brownNessuna valutazione finora
- Final IRA - Mangoes From IndiaDocumento174 pagineFinal IRA - Mangoes From Indiasakthi_dtsNessuna valutazione finora
- Gene InteractionDocumento23 pagineGene Interactionwillayat aliNessuna valutazione finora
- A Survey of Color Charts For Biological DescriptionsDocumento14 pagineA Survey of Color Charts For Biological DescriptionsgermporeNessuna valutazione finora
- Cointegrated VectorsDocumento2 pagineCointegrated Vectorsdevikamurugan124206Nessuna valutazione finora
- PG I IflashDocumento4 paginePG I IflashNIGHT tubeNessuna valutazione finora
- 8 13 PDFDocumento7 pagine8 13 PDFVillycia LNessuna valutazione finora