Potrebbero piacerti anche
- Os segredos para estudar e se preparar para provasDocumento114 pagineOs segredos para estudar e se preparar para provasVinícius Morais BredaNessuna valutazione finora
- ValidaçãoDocumento44 pagineValidaçãoRafa Oliveira100% (2)
- Projeto de lavador Venturi para remoção de particulado em processo de produção de fertilizanteDocumento8 pagineProjeto de lavador Venturi para remoção de particulado em processo de produção de fertilizanteAmauri HenriqueNessuna valutazione finora
- Orientações Para O Uso De Fitoterápicos E Plantas MedicinaisDa EverandOrientações Para O Uso De Fitoterápicos E Plantas MedicinaisNessuna valutazione finora
- Registro e Pos Registro de MedicamentosDocumento74 pagineRegistro e Pos Registro de MedicamentosDiego Marcelo100% (1)
- LISTA DE EXERCÍCIOS - FUNÇÕES OXIGENADAS R - 2º TRIMESTREDocumento4 pagineLISTA DE EXERCÍCIOS - FUNÇÕES OXIGENADAS R - 2º TRIMESTREkakayolimaNessuna valutazione finora
- RDC 166/2017 estabelece parâmetros validação métodos análiseDocumento2 pagineRDC 166/2017 estabelece parâmetros validação métodos análiseGabriela ZátteraNessuna valutazione finora
- Legislação brasileira sobre registro de fitoterápicosDocumento27 pagineLegislação brasileira sobre registro de fitoterápicosJoedi Santos100% (1)
- GASESDocumento39 pagineGASESJüvinal Pasiensia Simu DeitNessuna valutazione finora
- Micro Biolog I ADocumento26 pagineMicro Biolog I ARogerio CastroNessuna valutazione finora
- Microbiologia e Imunologia BacterianaDocumento44 pagineMicrobiologia e Imunologia BacterianaTarcizio FilhoNessuna valutazione finora
- Processo de Producao de Molho de Tomate Reator Bateladas PDFDocumento144 pagineProcesso de Producao de Molho de Tomate Reator Bateladas PDFGustavo Ruas100% (2)
- RDC 16-2007Documento10 pagineRDC 16-2007brunagabrielarochaNessuna valutazione finora
- RDC 17-07Documento12 pagineRDC 17-07donato_synchrNessuna valutazione finora
- Resolução ANVS 391.99Documento35 pagineResolução ANVS 391.99tielecarvalhoNessuna valutazione finora
- Procedimento simplificado registro medicamentosDocumento6 pagineProcedimento simplificado registro medicamentosLuis JibrinNessuna valutazione finora
- rdc0031 29 05 2014Documento10 paginerdc0031 29 05 2014Matheus Meireles de OliveiraNessuna valutazione finora
- RDC 35 2012 Medicamento ReferênciaDocumento11 pagineRDC 35 2012 Medicamento ReferênciaGabriel BiancoNessuna valutazione finora
- Critérios para registro de medicamentos sintéticos e semissintéticosDocumento22 pagineCritérios para registro de medicamentos sintéticos e semissintéticosArq ClayNessuna valutazione finora
- RDC 31 2014 CompDocumento13 pagineRDC 31 2014 CompGabriel MacedoNessuna valutazione finora
- RDC No 60 estabelece critérios para registro de medicamentosDocumento18 pagineRDC No 60 estabelece critérios para registro de medicamentospriflorindaNessuna valutazione finora
- RDC 200 2017 CompDocumento24 pagineRDC 200 2017 CompPedro ZanettiNessuna valutazione finora
- Regras para registro de fitoterápicosDocumento4 pagineRegras para registro de fitoterápicosRafaelaalmNessuna valutazione finora
- Requisitos para registro de medicamentos genéricos e similaresDocumento7 pagineRequisitos para registro de medicamentos genéricos e similaresEmilly EmanueleNessuna valutazione finora
- (2015) Consolidado - COFID - VDocumento737 pagine(2015) Consolidado - COFID - VflaviaNessuna valutazione finora
- Estudo DirigidoDocumento3 pagineEstudo DirigidoRafaela AlmeidaNessuna valutazione finora
- Resolução #4, de 10 de Fevereiro de 2009Documento6 pagineResolução #4, de 10 de Fevereiro de 2009Kleberson SouzaNessuna valutazione finora
- Medicamentos Similares PDFDocumento26 pagineMedicamentos Similares PDFJoéder AraújoNessuna valutazione finora
- ANVISA Resolução RDC N 333Documento44 pagineANVISA Resolução RDC N 333Rachel GarciaNessuna valutazione finora
- RDC 98 2016Documento9 pagineRDC 98 2016haphaeoNessuna valutazione finora
- RDC 23 1999 CompDocumento6 pagineRDC 23 1999 CompMaria Julia BistricanNessuna valutazione finora
- RDC 31 2010 CompDocumento17 pagineRDC 31 2010 CompRafael NunesNessuna valutazione finora
- Aula 2 - Legislação Sobre Medicamentos Referência, Genéricos e SimilaresDocumento9 pagineAula 2 - Legislação Sobre Medicamentos Referência, Genéricos e SimilaresmagdalaneferreiraNessuna valutazione finora
- Medicamentos SimilaresDocumento11 pagineMedicamentos SimilaresPamela Honorato100% (1)
- Consolidado de normas COFIDDocumento19 pagineConsolidado de normas COFIDFernanda Sacramento PeixotoNessuna valutazione finora
- Regras para medicamentos isentos de prescriçãoDocumento9 pagineRegras para medicamentos isentos de prescriçãomarlyton marquesNessuna valutazione finora
- Presidência Da República: Casa CivilDocumento3 paginePresidência Da República: Casa CivilElviradeMelloNessuna valutazione finora
- Medicamentos genéricos x referência x similares: diferenças e regulamentaçãoDocumento13 pagineMedicamentos genéricos x referência x similares: diferenças e regulamentaçãoJanaina FernandesNessuna valutazione finora
- Boas Práticas FarmacêuticasDocumento30 pagineBoas Práticas Farmacêuticasgbrandao8Nessuna valutazione finora
- Resolução - RDC #20, de 5 de Maio de 2011: Minist Rio Da Sa deDocumento5 pagineResolução - RDC #20, de 5 de Maio de 2011: Minist Rio Da Sa deSamanta PontesNessuna valutazione finora
- RDC 31Documento9 pagineRDC 31Laryssa AssisNessuna valutazione finora
- Regulamenta registro medicamentos específicosDocumento5 pagineRegulamenta registro medicamentos específicosPaulo BeneventoNessuna valutazione finora
- Lei Dos Genéricos 9787-99Documento3 pagineLei Dos Genéricos 9787-99Aramís PereiraNessuna valutazione finora
- Conceitos Técnicos ANVISADocumento11 pagineConceitos Técnicos ANVISAIANA HAASNessuna valutazione finora
- Consolidado Fitoterapicos 2018Documento655 pagineConsolidado Fitoterapicos 2018Vanderson LimaNessuna valutazione finora
- Retropecto Dos Marcos Legais Adotados Pela Anvisa para o Registro de Medicamentos Genéricos e SimilaresDocumento21 pagineRetropecto Dos Marcos Legais Adotados Pela Anvisa para o Registro de Medicamentos Genéricos e SimilaresCláudia Maria RangelNessuna valutazione finora
- Nota Técnica Nº 17 de 2015 - COGENDocumento4 pagineNota Técnica Nº 17 de 2015 - COGENGabriel MacedoNessuna valutazione finora
- RDC 37Documento11 pagineRDC 37Joao DinizNessuna valutazione finora
- RDC 67 AnexoDocumento101 pagineRDC 67 AnexowillchernobylNessuna valutazione finora
- Atividade FHDocumento12 pagineAtividade FHAryanne AlvesNessuna valutazione finora
- Prescrição e dispensação de medicamentos genéricos e similares segundo a legislação brasileiraDocumento5 paginePrescrição e dispensação de medicamentos genéricos e similares segundo a legislação brasileiraRodrigo LeiteNessuna valutazione finora
- RDC 33 2000Documento41 pagineRDC 33 2000Fabinho GarciaNessuna valutazione finora
- Relatorio Farmacia ComunitáriaDocumento12 pagineRelatorio Farmacia ComunitáriaJessyka AbreuNessuna valutazione finora
- RDC #753Documento45 pagineRDC #753brunaemilialimaNessuna valutazione finora
- RDC 31 2010Documento8 pagineRDC 31 2010LUCAS DOS SANTOS NUNESNessuna valutazione finora
- Regulamentação da fitoterapia no BrasilDocumento10 pagineRegulamentação da fitoterapia no Brasilleticia0% (1)
- Tabela Toxicidade AnvisaDocumento31 pagineTabela Toxicidade AnvisaPatricia souzaNessuna valutazione finora
- RDC 71 09Documento19 pagineRDC 71 09Thiago LimaNessuna valutazione finora
- Registro UnifsmDocumento5 pagineRegistro UnifsmRaimundo Fernandes Leite NetoNessuna valutazione finora
- Registro de medicamentos no Instituto Politécnico Agrário de MaputoDocumento11 pagineRegistro de medicamentos no Instituto Politécnico Agrário de MaputoOsvaldo GuilimaNessuna valutazione finora
- FarmaciaDocumento11 pagineFarmaciaOsvaldo GuilimaNessuna valutazione finora
- Arquivo 2022EBookComoDescobrirseumprodutoDocumento37 pagineArquivo 2022EBookComoDescobrirseumprodutoFrancine Mendes ReisNessuna valutazione finora
- RDC 60 2009 - Amostras GratisDocumento3 pagineRDC 60 2009 - Amostras GratisLydi LoboNessuna valutazione finora
- RDC 67 2007 - Água Purificada (Farmácia de Manipulação) PDFDocumento90 pagineRDC 67 2007 - Água Purificada (Farmácia de Manipulação) PDFJefferson LinsNessuna valutazione finora
- RDC 471 estabelece critérios para prescrição e dispensação de antimicrobianosDocumento5 pagineRDC 471 estabelece critérios para prescrição e dispensação de antimicrobianosviniciusNessuna valutazione finora
- Lista preços medicamentosDocumento13 pagineLista preços medicamentosRivaldo SantosNessuna valutazione finora
- Anvisa glossário definições medicamentosDocumento6 pagineAnvisa glossário definições medicamentosCarlos Diego Quirino LimaNessuna valutazione finora
- Registro de Medicamentos AnvisaDocumento49 pagineRegistro de Medicamentos AnvisaRogerio CastroNessuna valutazione finora
- 46 61 SalaslimpasDocumento16 pagine46 61 SalaslimpasRogerio CastroNessuna valutazione finora
- MEDICAMENTOSDocumento1 paginaMEDICAMENTOSRogerio CastroNessuna valutazione finora
- Resolucao 338 Politica Ass FarmaceuticaDocumento2 pagineResolucao 338 Politica Ass FarmaceuticaEduardo FrizzeraNessuna valutazione finora
- Os Princípios Do SUS - SUS Princípios e ConquistasDocumento43 pagineOs Princípios Do SUS - SUS Princípios e ConquistasWagner AlvesNessuna valutazione finora
- Minitestes Biologia CelularDocumento9 pagineMinitestes Biologia CelularJOAO FRANCISCONessuna valutazione finora
- 6 Pop Fracionamento de Medicamentos PDFDocumento4 pagine6 Pop Fracionamento de Medicamentos PDFSandro PinheiroNessuna valutazione finora
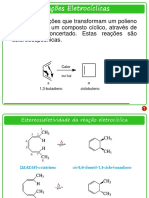
- Reações Eletrocíclicas e Rearranjos SigmatrópicosDocumento24 pagineReações Eletrocíclicas e Rearranjos SigmatrópicosAnonymous UYDJtUnNessuna valutazione finora
- Resistencia Ao FogoDocumento16 pagineResistencia Ao FogolubakyNessuna valutazione finora
- Ensaios de CorrosãoDocumento4 pagineEnsaios de CorrosãosergiodelbiancofilhoNessuna valutazione finora
- Aula 1 - Propriedades Físicas Dos Fluidos e Sistemas de UnidadesDocumento16 pagineAula 1 - Propriedades Físicas Dos Fluidos e Sistemas de UnidadesvicentenetokindleNessuna valutazione finora
- Manual do Usuário Aquecedor a Gás KOMECODocumento28 pagineManual do Usuário Aquecedor a Gás KOMECOLuizNessuna valutazione finora
- O que é um átomo e como é sua estruturaDocumento50 pagineO que é um átomo e como é sua estruturaMarcelo Mendes SantucciNessuna valutazione finora
- Teste 9ºano CN 2 CorrecçãoDocumento4 pagineTeste 9ºano CN 2 CorrecçãoRita CardosoNessuna valutazione finora
- Medicina Química Termoquímica Mistura Dos Casos Exercícios Específicos 19-07-2018Documento15 pagineMedicina Química Termoquímica Mistura Dos Casos Exercícios Específicos 19-07-2018belinhaNessuna valutazione finora
- Células e Material GenéticoDocumento15 pagineCélulas e Material GenéticoNatane S. GomesNessuna valutazione finora
- Problemas Intrínsecos e Graves Da Expansão Mineral, Metalúrgica, Petrolífera, e Hidrelétrica Nas Amazônias - A. Oswaldo Sevá FoDocumento28 pagineProblemas Intrínsecos e Graves Da Expansão Mineral, Metalúrgica, Petrolífera, e Hidrelétrica Nas Amazônias - A. Oswaldo Sevá FoVivian PradoNessuna valutazione finora
- Aterro Industrial: Funcionamento e RequisitosDocumento16 pagineAterro Industrial: Funcionamento e RequisitosRoberta Figueira SilvaNessuna valutazione finora
- Caracterização de CarbetosDocumento10 pagineCaracterização de Carbetosz DrakNessuna valutazione finora
- Lâmpadas LED OSRAM: vantagens e portfólio completoDocumento35 pagineLâmpadas LED OSRAM: vantagens e portfólio completoludmilaNessuna valutazione finora
- Processos de produção de aço em usinas siderúrgicasDocumento210 pagineProcessos de produção de aço em usinas siderúrgicasDanilo HoskenNessuna valutazione finora
- Veja Banheiro Com Cloro Ativo - Fispq Rev03Documento9 pagineVeja Banheiro Com Cloro Ativo - Fispq Rev03Ramon Teixeira DiasNessuna valutazione finora
- Sistema Internacional de Unidades e outras unidades de medidaDocumento15 pagineSistema Internacional de Unidades e outras unidades de medidaBruno CunhaNessuna valutazione finora
- Execução e Acabamento Do Concreto Projetado - Especificação de Serviço.Documento11 pagineExecução e Acabamento Do Concreto Projetado - Especificação de Serviço.mdrygallaNessuna valutazione finora
- Avaliação de Geografia sobre a Europa DesenvolvidaDocumento3 pagineAvaliação de Geografia sobre a Europa DesenvolvidaEtevaldo LimaNessuna valutazione finora
- TrabalhoDocumento4 pagineTrabalhoVictoria Rabelo FerrazNessuna valutazione finora
- Manual Proc Fabr IIDocumento58 pagineManual Proc Fabr IIAdam IsmailNessuna valutazione finora
- Atriplex nova forrageira solos salinizados NEDocumento2 pagineAtriplex nova forrageira solos salinizados NEhoracioarsNessuna valutazione finora