Potrebbero piacerti anche
- Diagnosis of Arteriovenous Malformation With Color Doppler UltrasoundDocumento2 pagineDiagnosis of Arteriovenous Malformation With Color Doppler UltrasoundKurnia Sari SyaifulNessuna valutazione finora
- Tag APS Test Laboratory PDFDocumento1 paginaTag APS Test Laboratory PDFKurnia Sari SyaifulNessuna valutazione finora
- Vascular Endothelial Growth Factor Expression in The Endometrium During The Menstrual CycleDocumento1 paginaVascular Endothelial Growth Factor Expression in The Endometrium During The Menstrual CycleKurnia Sari SyaifulNessuna valutazione finora
- Antiphospolipid Syndrome PDFDocumento9 pagineAntiphospolipid Syndrome PDFKurnia Sari SyaifulNessuna valutazione finora
- Case Report AVM FIX ENGLISH!!Documento10 pagineCase Report AVM FIX ENGLISH!!Kurnia Sari SyaifulNessuna valutazione finora
- Case KET ALDocumento31 pagineCase KET ALKurnia Sari SyaifulNessuna valutazione finora
- Intrapartum Complication of MacrosomiaDocumento4 pagineIntrapartum Complication of MacrosomiaKurnia Sari SyaifulNessuna valutazione finora
- Eklampsi Antepartum Hellp Syndrom + DIC + IctericDocumento32 pagineEklampsi Antepartum Hellp Syndrom + DIC + IctericKurnia Sari SyaifulNessuna valutazione finora
- Plasenta PreviaDocumento21 paginePlasenta PreviaKurnia Sari SyaifulNessuna valutazione finora
- 191 Acute Fatty Liver of PregnancyDocumento5 pagine191 Acute Fatty Liver of PregnancyKurnia Sari SyaifulNessuna valutazione finora
- Culdocentesis and Ectopic PDFDocumento1 paginaCuldocentesis and Ectopic PDFKurnia Sari SyaifulNessuna valutazione finora
- Culdocentesis and Ectopic PDFDocumento1 paginaCuldocentesis and Ectopic PDFKurnia Sari SyaifulNessuna valutazione finora
- Ectopic PregnancyDocumento3 pagineEctopic PregnancyKurnia Sari SyaifulNessuna valutazione finora
- OBG028 - Abnormal Uterine Bleeding Uterine Fibroids - 12-12 PDFDocumento23 pagineOBG028 - Abnormal Uterine Bleeding Uterine Fibroids - 12-12 PDFKurnia Sari SyaifulNessuna valutazione finora
- Liver Disease With PregnancyDocumento9 pagineLiver Disease With PregnancyKurnia Sari SyaifulNessuna valutazione finora
- Culdocentesis and Ectopic PDFDocumento1 paginaCuldocentesis and Ectopic PDFKurnia Sari SyaifulNessuna valutazione finora
- Minimizing The Occurence and Complications of PreeclampsiaDocumento57 pagineMinimizing The Occurence and Complications of PreeclampsiaKurnia Sari SyaifulNessuna valutazione finora
- English CaseDocumento3 pagineEnglish CaseKurnia Sari SyaifulNessuna valutazione finora
- 191 Acute Fatty Liver of PregnancyDocumento5 pagine191 Acute Fatty Liver of PregnancyKurnia Sari SyaifulNessuna valutazione finora
- Obesity and Infertility: Behind The Medical HeadlinesDocumento4 pagineObesity and Infertility: Behind The Medical HeadlinesKurnia Sari SyaifulNessuna valutazione finora
- Universal PrecautionDocumento48 pagineUniversal PrecautionThirugnanaThiru100% (1)
- Antiphospholipid SyndromeDocumento12 pagineAntiphospholipid SyndromeKurnia Sari SyaifulNessuna valutazione finora
- Clinical Significance of Anticardiolipin and Anti B2GPI Antibodies PDFDocumento7 pagineClinical Significance of Anticardiolipin and Anti B2GPI Antibodies PDFKurnia Sari SyaifulNessuna valutazione finora
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (73)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- University of Cambridge International Examinations General Certificate of Education Ordinary LevelDocumento16 pagineUniversity of Cambridge International Examinations General Certificate of Education Ordinary Levelmstudy123456Nessuna valutazione finora
- 1 Diabetes Screening PO4047576719 696Documento4 pagine1 Diabetes Screening PO4047576719 696rishiranjan04Nessuna valutazione finora
- Antritrombina IIIDocumento8 pagineAntritrombina IIIisabel bautistaNessuna valutazione finora
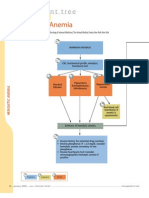
- Diagnostic Tree - Hemolytic AnemiaDocumento2 pagineDiagnostic Tree - Hemolytic AnemiaЂорђеNessuna valutazione finora
- IGCSE Biology NotesDocumento15 pagineIGCSE Biology NotesCrystal Wong100% (14)
- Signs of VTE InfographicDocumento1 paginaSigns of VTE InfographicJulia Martha LinardiNessuna valutazione finora
- Quiz Cell 3oktDocumento25 pagineQuiz Cell 3oktFarah AsnidaNessuna valutazione finora
- Coagulometre Bi-CannauxDocumento4 pagineCoagulometre Bi-Cannauxamor kermayaNessuna valutazione finora
- Breathing & Exchange of GasesDocumento5 pagineBreathing & Exchange of GasesMamta TiwariNessuna valutazione finora
- Physiological Effects of MassageDocumento1 paginaPhysiological Effects of MassagehannahNessuna valutazione finora
- Case Report: Immune Thrombocytopenic PurpuraDocumento35 pagineCase Report: Immune Thrombocytopenic PurpuraFelinNessuna valutazione finora
- Blood Typing PresentationDocumento39 pagineBlood Typing PresentationNezzeh Clair D. BermejoNessuna valutazione finora
- PROVA - New Fce Test 000 ReadingDocumento5 paginePROVA - New Fce Test 000 ReadingLuís Fernando ZaltronNessuna valutazione finora
- TRANSFUSION Basic Guidelines Principles and Practice 2011Documento52 pagineTRANSFUSION Basic Guidelines Principles and Practice 2011entistdeNessuna valutazione finora
- Transport in Mammals: The Need For A Transport SystemDocumento2 pagineTransport in Mammals: The Need For A Transport SystemAhmed OmarNessuna valutazione finora
- C72IA025EN-B Viscosity of Whole BloodDocumento3 pagineC72IA025EN-B Viscosity of Whole BloodMartín PerezNessuna valutazione finora
- Tao of Forgotten Food Diet - Taoist HerbologyDocumento27 pagineTao of Forgotten Food Diet - Taoist HerbologyZioAngel100% (1)
- Why Are You Wearing Gold Jewellery, Amazing Facts of Gold Jewellery, Science of Gold Jewellery, Philosphy of Gold JewelleryDocumento43 pagineWhy Are You Wearing Gold Jewellery, Amazing Facts of Gold Jewellery, Science of Gold Jewellery, Philosphy of Gold JewelleryRaghu.GNessuna valutazione finora
- Biology Form 4 Chapter 2Documento23 pagineBiology Form 4 Chapter 2Thiyaku MaruthaNessuna valutazione finora
- Top 5 Causes of Splenomegaly in Dogs - Clinicians BriefDocumento5 pagineTop 5 Causes of Splenomegaly in Dogs - Clinicians Briefludiegues752Nessuna valutazione finora
- Mark Scheme (Results) January 2013: International GCSE Biology (4BI0) Paper 1BDocumento20 pagineMark Scheme (Results) January 2013: International GCSE Biology (4BI0) Paper 1BJohn HopkinsNessuna valutazione finora
- DiabetesDocumento1 paginaDiabetesHartford CourantNessuna valutazione finora
- Directions: Each Question Below Contains Five Suggested Answers. Choose The One Best ResponseDocumento4 pagineDirections: Each Question Below Contains Five Suggested Answers. Choose The One Best ResponseTrottTamilNessuna valutazione finora
- CC CM Isbb Histopath Mtlaw ReviewerDocumento26 pagineCC CM Isbb Histopath Mtlaw ReviewerAnne MorenoNessuna valutazione finora
- Session #27 SAS - AnaPhy (Lab)Documento4 pagineSession #27 SAS - AnaPhy (Lab)G INessuna valutazione finora
- LECTURE 6 The ABO Blood Group SystemDocumento64 pagineLECTURE 6 The ABO Blood Group SystemKrisyah Niqoule Valdez100% (2)
- Homeostasis PosterDocumento1 paginaHomeostasis Posterapi-375897947Nessuna valutazione finora
- CicaLiquid NH3 - L-020EDocumento4 pagineCicaLiquid NH3 - L-020Eandr sonNessuna valutazione finora
- Normal Laboratory ValuesDocumento4 pagineNormal Laboratory ValuesPaulene CartelNessuna valutazione finora
- NEO Scavenger - Manual PDFDocumento37 pagineNEO Scavenger - Manual PDFFilha Do ReiNessuna valutazione finora