Potrebbero piacerti anche
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
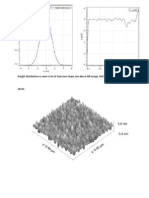
- Height Distribution Is Seen To Be of Gaussian Shape See Above Left Image. HHCF Fit On The RightDocumento2 pagineHeight Distribution Is Seen To Be of Gaussian Shape See Above Left Image. HHCF Fit On The Rightkevers_keversNessuna valutazione finora
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- FGBRDocumento15 pagineFGBRkevers_keversNessuna valutazione finora
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5794)
- FGBRDocumento15 pagineFGBRkevers_keversNessuna valutazione finora
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (399)
- Lang Code MybbDocumento3 pagineLang Code Mybbwhite_snow19Nessuna valutazione finora
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- Lang Code MybbDocumento3 pagineLang Code Mybbwhite_snow19Nessuna valutazione finora
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- Raswry PhysDocumento1 paginaRaswry Physkevers_keversNessuna valutazione finora
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- Metaphysics of LucretiusDocumento6 pagineMetaphysics of LucretiusChristopher BennettNessuna valutazione finora
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- Minimalist KWL Graphic OrganizerDocumento2 pagineMinimalist KWL Graphic OrganizerIrish Nicole AlanoNessuna valutazione finora
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- Resume: Satyam KumarDocumento3 pagineResume: Satyam KumarEr Satyam Kumar KrantiNessuna valutazione finora
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- Contoh CV / Daftar Riwayat HidupDocumento2 pagineContoh CV / Daftar Riwayat HiduprusmansyahNessuna valutazione finora
- Investigation of Skew Curved Bridges in Combination With Skewed Abutments Under Seismic ResponseDocumento5 pagineInvestigation of Skew Curved Bridges in Combination With Skewed Abutments Under Seismic ResponseEditor IJTSRDNessuna valutazione finora
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- Integration ConceptDocumento34 pagineIntegration ConceptJANELLA ALVAREZNessuna valutazione finora
- Dairy Products Theory XIIDocumento152 pagineDairy Products Theory XIIDskNessuna valutazione finora
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- Gondola CalculationDocumento6 pagineGondola CalculationBudi SusantoNessuna valutazione finora
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (73)
- Harmonic Analysis of Separately Excited DC Motor Drives Fed by Single Phase Controlled Rectifier and PWM RectifierDocumento112 pagineHarmonic Analysis of Separately Excited DC Motor Drives Fed by Single Phase Controlled Rectifier and PWM RectifierGautam Umapathy0% (1)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (344)
- FebvreDocumento449 pagineFebvreIan Pereira AlvesNessuna valutazione finora
- ASME Pressue Vessel ExampleDocumento271 pagineASME Pressue Vessel ExamplesavanchandranNessuna valutazione finora
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- CCNA Training New CCNA - RSTPDocumento7 pagineCCNA Training New CCNA - RSTPokotete evidenceNessuna valutazione finora
- Pusheen With Donut: Light Grey, Dark Grey, Brown, RoséDocumento13 paginePusheen With Donut: Light Grey, Dark Grey, Brown, RosémafaldasNessuna valutazione finora
- Pharmalytica Exhibitor List 2023Documento3 paginePharmalytica Exhibitor List 2023Suchita PoojaryNessuna valutazione finora
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- YoungMan EN131 GUIDEDocumento16 pagineYoungMan EN131 GUIDErcpawar100% (1)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- Anderson, Poul - Flandry 02 - A Circus of HellsDocumento110 pagineAnderson, Poul - Flandry 02 - A Circus of Hellsgosai83Nessuna valutazione finora
- Esterification Oil of WintergreenDocumento8 pagineEsterification Oil of WintergreenMaria MahusayNessuna valutazione finora
- Surface Finish Measurement NotesDocumento32 pagineSurface Finish Measurement NotesAneez ShresthaNessuna valutazione finora
- The Indian & The SnakeDocumento3 pagineThe Indian & The SnakeashvinNessuna valutazione finora
- MA 106: Linear Algebra Tutorial 1: Prof. B.V. Limaye IIT DharwadDocumento4 pagineMA 106: Linear Algebra Tutorial 1: Prof. B.V. Limaye IIT Dharwadamar BaroniaNessuna valutazione finora
- DIVAR IP All-In-One 7000 3U Datasheet 51 en 66297110155Documento5 pagineDIVAR IP All-In-One 7000 3U Datasheet 51 en 66297110155Javier RochaNessuna valutazione finora
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- English Class Vii PDFDocumento101 pagineEnglish Class Vii PDFpannapurohitNessuna valutazione finora
- Line Differential Protection Red670Documento8 pagineLine Differential Protection Red670igorsfaceNessuna valutazione finora
- Maintenance Páginas-509-580Documento72 pagineMaintenance Páginas-509-580Alexandra Gabriela Pacheco PrietoNessuna valutazione finora
- Data SiEMEx School SafetyPreparedness 25 26 NOVDocumento81 pagineData SiEMEx School SafetyPreparedness 25 26 NOVSuraj RajuNessuna valutazione finora
- Crma Unit 1 Crma RolesDocumento34 pagineCrma Unit 1 Crma Rolesumop3plsdn0% (1)
- 2017 Classification of Periodontal and Peri-Implant Diseases and Conditions. Decision Making Algorithms For Clinical PracticeDocumento40 pagine2017 Classification of Periodontal and Peri-Implant Diseases and Conditions. Decision Making Algorithms For Clinical PracticebbNessuna valutazione finora
- DP November 2017 Examination Schedule en PDFDocumento4 pagineDP November 2017 Examination Schedule en PDFSuperlucidoNessuna valutazione finora
- V. Jovicic and M. R. Coop1997 - Stiffness, Coarse Grained Soils, Small StrainsDocumento17 pagineV. Jovicic and M. R. Coop1997 - Stiffness, Coarse Grained Soils, Small StrainsxiangyugeotechNessuna valutazione finora
- Cold Regions Science and TechnologyDocumento8 pagineCold Regions Science and TechnologyAbraham SilesNessuna valutazione finora
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)