Documenti di Didattica
Documenti di Professioni
Documenti di Cultura
Instrumental Methods of Analysis Lesson Notes
Caricato da
coolhemakumar0 valutazioniIl 0% ha trovato utile questo documento (0 voti)
65 visualizzazioni80 pagineA quantitative method provides numerical inIormation as to the relative amount oI one or more oI these components in the sample. Instrumentation can be divided into two categories: detection and quantitation. A qualitative method yields information about the identity oI atomic or molecular species.
Descrizione originale:
Copyright
© Attribution Non-Commercial (BY-NC)
Formati disponibili
PDF, TXT o leggi online da Scribd
Condividi questo documento
Condividi o incorpora il documento
Hai trovato utile questo documento?
Questo contenuto è inappropriato?
Segnala questo documentoA quantitative method provides numerical inIormation as to the relative amount oI one or more oI these components in the sample. Instrumentation can be divided into two categories: detection and quantitation. A qualitative method yields information about the identity oI atomic or molecular species.
Copyright:
Attribution Non-Commercial (BY-NC)
Formati disponibili
Scarica in formato PDF, TXT o leggi online su Scribd
0 valutazioniIl 0% ha trovato utile questo documento (0 voti)
65 visualizzazioni80 pagineInstrumental Methods of Analysis Lesson Notes
Caricato da
coolhemakumarA quantitative method provides numerical inIormation as to the relative amount oI one or more oI these components in the sample. Instrumentation can be divided into two categories: detection and quantitation. A qualitative method yields information about the identity oI atomic or molecular species.
Copyright:
Attribution Non-Commercial (BY-NC)
Formati disponibili
Scarica in formato PDF, TXT o leggi online su Scribd
Sei sulla pagina 1di 80
UNIT I BASICS OF MEASUREMENT
ClassiIication oI methods calibration oI instrumental methods electrical
components and circuits signal to noise ratio signal noise enhancement.
INTRODUCTION
Analytical Chemistry deals with methods Ior determining the chemical composition
oI samples oI matter. A qualitative method yields inIormation about the identity oI atomic or
molecular species or the Iunctional groups in the sample; a quantitative method, in contrast,
provides numerical inIormation as to the relative amount oI one or more oI these
components.
Analytical methods are oIten classiIied as being either classical or instrumental. This
classiIication is largely historical with classical methods, sometimes called wet-chemical
methods, preceding instrumental methods by a century or more.
Classical Methods
Separation oI analytes by precipitation, extraction, or distillation.
Qualitative analysis by reaction oI analytes with reagents that yielded products that
could be recognized by their colors, boiling or melting points, solubilities, optical
activities, or reIractive indexes.
Quantitative analysis by gravimetric or by titrimetric techniques.
1. Gravimetric Methods the mass oI the analyte or some compound produced Irom the
analyte was determined.
2. Titrimetric Methods the volume or mass oI a standard reagent required to react
completely with the analyte was measured.
Instrumental Methods
Measurements oI physical properties oI analytes, such as conductivity, electrode
potential, light absorption, or emission, mass to charge ratio, and Iluorescence, began to be
used Ior quantitative analysis oI a variety oI inorganic, organic, and biochemical analyte.
Highly eIIicient chromatographic and electrophoretic techniques began to replace distillation,
extraction, and precipitation Ior the separation oI components oI complex mixtures prior to
their qualitative or quantitative determination. These newer methods Ior separating and
determining chemical species are known collectively as instrumental methods oI analysis.
Instrumentation can be divided into two categories: detection and quantitation.
1. Quantitation
Measurement oI physical properties oI analytes - such as conductivity, electrode
potential, light absorption or emission, mass-to-charge ratio, and Iluorescence-began to be
employed Ior quantitative analysis oI inorganic, organic, and biochemical analytes.
2. Detection
EIIicient chromatographic separation techniques are used Ior the separation oI
components oI complex mixtures.
Table 1. Classification of instrumental methods based on different analytical signals
Signal Instrumental Methods
Emission oI radiation Emission spectroscopy (X-ray, UV, visible,
electron, Auger); Iluorescence,
phosphorescence, and luminescence
(X-ray, UV, and visible)
Absorption oI radiation Spectrophotometry and photometry (X-ray,
UV, visible, IR); photoacoustic
spectroscopy; nuclear magnetic resonance
and electron spin resonance spectroscopy
Scattering oI radiation Turbidimetry; nephelometry; Raman
spectroscopy
ReIraction oI radiation ReIractometry; interIerometry
DiIIraction oI radiation X-Ray and electron diIIraction methods
Rotation oI radiation Polarimetry; optical rotary dispersion;
circular dichroism
Electrical potential Potentiometry; chronopotentiometry
Electrical charge Coulometry
Electrical current Polarography; amperometry
Electrical resistance Conductometry
Mass-to-charge ratio Mass spectrometry
Rate oI reaction Kinetic methods
Thermal properties Thermal conductivity and enthalpy
Radioactivity Activation and isotope dilution methods
CALIBRATION OF INSTRUMENTAL METHODS
A calibration curve is one approach to the problem oI instrument calibration; other
approaches may mix the standard into the unknown, giving an internal standard.
The calibration curve is a plot oI how the instrumental response, the so-called
analytical signal, changes with the concentration oI the analyte (the substance to be
measured). The operator prepares a series oI standards across a range oI concentrations near
the expected concentration oI analyte in the unknown. The concentrations oI the standards
must lie within the working range oI the technique (instrumentation) they are using (see
Iigure). Analyzing each oI these standards using the chosen technique will produce a series oI
measurements. For most analyses a plot oI instrument response vs. analyte concentration will
show a linear relationship. The operator can measure the response oI the unknown and, using
the calibration curve, can interpolate to Iind the concentration oI analyte.
Calibration curve
Figure 1.Limit oI detection (LOD), limit oI quantiIication (LOQ), dynamic range, and limit oI linearity (LOL).
How to create a calibration curve
The data - the concentrations oI the analyte and the instrument response Ior each
standard - can be Iit to a straight line, using linear regression analysis. This yields a model
described by the equation y mx + c, where y is the instrument response, m represents the
sensitivity, and c is a constant that describes the background. The analyte concentration (x) oI
unknown samples may be calculated Irom this equation.
Many diIIerent variables can be used as the analytical signal. For instance, chromium
(III) might be measured using a chemiluminescence method, in an instrument that contains a
photomultiplier tube (PMT) as the detector. The detector converts the light produced by the
sample into a voltage, which increases with intensity oI light. The amount oI light measured
is the analytical signal.
Most analytical techniques use a calibration curve. There are a number oI advantages
to this approach. First, the calibration curve provides a reliable way to calculate the
uncertainty oI the concentration calculated Irom the calibration curve (using the statistics oI
the least squares line Iit to the data).
|1|
Second, the calibration curve provides data on an empirical relationship. The
mechanism Ior the instrument's response to the analyte may be predicted or understood
according to some theoretical model, but most such models have limited value Ior real
samples. (Instrumental response is usually highly dependent on the condition oI the analyte,
solvents used and impurities it may contain; it could also be aIIected by external Iactors such
as pressure and temperature.)
Many theoretical relationships, such as Iluorescence, require the determination oI an
instrumental constant anyway, by analysis oI one or more reIerence standards; a calibration
curve is a convenient extension oI this approach. The calibration curve Ior a particular
analyte in a particular (type oI) sample provides the empirical relationship needed Ior those
particular measurements.
The chieI disadvantages are that the standards require a supply oI the analyte material,
preIerably oI high purity and in known concentration. (Some analytes - e.g., particular
proteins - are extremely diIIicult to obtain pure in suIIicient quantity.)
Applications
Analysis oI concentration
VeriIying the proper Iunctioning oI an analytical instrument or a sensor device such as an
ion selective electrode
Determining the basic eIIects oI a control treatment (such as a dose-survival curve in
clonogenic assay)
Standard addition method
The method oI standard addition is used in instrumental analysis to determine
concentration oI a substance (analyte) in an unknown sample by comparison to a set oI
samples oI known concentration, similar to using a calibration curve. Standard addition can
be applied to most analytical techniques and is used instead oI a calibration curve to solve the
matrix eIIect problem.
This graph is an example oI a standard addition plot used to determine the
concentration oI calcium in an unknown sample by atomic absorption spectroscopy. The
point at zero concentration added Ca is the reading oI the unknown, the other points are the
readings aIter adding increasing amounts ('spikes') oI standard solution. The absolute value
of the x-intercept is the concentration oI Ca in the unknown, in this case 1.69E-6 g/mL.
Applications
Standard addition is Irequently used in atomic absorption spectroscopy and gas
chromatography.
LAWS OF ELECTRICITY
Ohm's Law
For many conductors oI electricity, the electric current which will Ilow through them
is directly proportional to the voltage applied to them. When a microscopic view oI Ohm's
law is taken, it is Iound to depend upon the Iact that the driIt velocity oI charges through the
material is proportional to the electric Iield in the conductor. The ratio oI voltage to current is
called the resistance, and iI the ratio is constant over a wide range oI voltages, the material is
said to be an "ohmic" material. II the material can be characterized by such a resistance, then
the current can be predicted Irom the relationship:
Kirchhoff's Current Law (KCL)
The current entering any junction is equal to the current leaving that junction. i
1
i
4
i
2
i
3.
This law is also called KirchhoII's Iirst law, KirchhoII's point rule, KirchhoII's junction
rule (or nodal rule), and KirchhoII's Iirst rule.
Power law
The general Iorm oI the law is
PVI
where I is the magnitude oI the physical stimulus, (I) is the psychophysical Iunction relating
to the subjective magnitude oI the sensation evoked by the stimulus, a is an exponent that
depends on the type oI stimulation and k is a proportionality constant that depends on the type
oI stimulation and the units used.
DIRECT CURRENT CIRCUITS AND MEASUREMENTS
Direct Current.(DC) Is one that Ilows always in the same direction. Most electronic
devices need Direct Current because they require a steady Ilow oI electrons that always head
in the same direction. A battery is Direct Current. Alternating Current (AC) is changed to
Direct Current (DC) with the use oI Diode RectiIiers. You cannot use a transIormer with
Direct Current.
Series Circuit
A Series circuit is one in which all components are connected in tandem. The current
at every point oI a series circuit stays the same. In series circuits the current remains the same
but the voltage drops may vary.
Parallel Circuit
Parallel circuits are those in which the components are so arranged that the current
divides between them. In parallel circuits the voltage remains the same but the current may
vary. The circuits in your home are wired in parallel.
MEASUREMENT OF DC
Digital voltmeters (DJM)
Digital voltmeters usually employ an electronic circuit that acts as an integrator,
linearly ramping output voltage when input voltage is constant (this can be easily
realized with an opamp). The dual-slope integrator method applies a known reIerence
voltage to the integrator Ior a Iixed time to ramp the integrator's output voltage up, then
the unknown voltage is applied to ramp it back down, and the time to ramp output
voltage down to zero is recorded (realized in an ADC implementation). The unknown
voltage being measured is the product oI the voltage reIerence and the ramp-up time
divided by the ramp-down time. The voltage reIerence must remain constant during the
ramp-up time, which may be diIIicult due to supply voltage and temperature variations.
Part oI the problem oI making an accurate voltmeter is that oI calibration to check its
accuracy. In laboratories, the Weston Cell is used as a standard voltage Ior precision
work. Precision voltage reIerences are available based on electronic circuits.Digital
voltmeters, like vacuum tube voltmeters, generally exhibit a constant input resistance oI
10 megohms regardless oI set measurement range.
ALTERNATING CURRENT CIRCUITS
The amplitude or peak value oI the sinusoidal variation we shall represent by V
m
and
I
m
, and we shall use V V
m
/2
1/2
and I I
m
/2
1/2
without subscripts to reIer to the RMS values.
For an explanation oI RMS values, see Power and RMS values.
So Ior instance, we shall write:
v v(t) V
m
sin (et )
i i(t) I
m
sin (et).
where e is the angular Irequency. e 2aI, where I is the ordinary or cyclic Irequency. I is the
number oI complete oscillations per second. is the phase diIIerence between the voltage
and current. We shall meet this and the geometrical signiIicance oI e later.
Resistors and Ohm's law in AC circuits
The voltage v across a resistor is proportional to the current i travelling through it.
Further, this is true at all times: v Ri. So, iI the current in a resistor is
i I
m
. sin (et) , we write:
v R.i R.I
m
sin (et)
v V
m
. sin (et) where
V
m
R.I
m
So Ior a resistor, the peak value oI voltage is R times the peak value oI current. Further, they
are in phase: when the current is a maximum, the voltage is also a maximum.
(Mathematically, 0.) The Iirst animation shows the voltage and current in a resistor as a
Iunction oI time.
Impedance and reactance
Circuits in which current is proportional to voltage are called linear circuits. (As soon
as one inserts diodes and transistors, circuits cease to be linear, but that's another story.) The
ratio oI voltage to current in a resistor is its resistance. Resistance does not depend on
Irequency, and in resistors the two are in phase, as we have seen in the animation. However,
circuits with only resistors are not very interesting.
In general, the ratio oI voltage to current does depend on Irequency and in general there is a
phase diIIerence. So impedance is the general name we give to the ratio oI voltage to current.
It has the symbol Z. Resistance is a special case oI impedance. Another special case is that in
which the voltage and current are out oI phase by 90: this is an important case because when
this happens, no power is lost in the circuit. In this case where the voltage and current are out
oI phase by 90, the ratio oI voltage to current is called the reactance, and it has the symbol
X.
Capacitors and charging
The voltage on a capacitor depends on the amount oI charge you store on its plates.
The current Ilowing onto the positive capacitor plate (equal to that Ilowing oII the negative
plate) is by deIinition the rate at which charge is being stored. So the charge Q on the
capacitor equals the integral oI the current with respect to time. From the deIinition oI the
capacitance,
v
C
q/C, so
we have a sinusoidal current i I
m
. sin (et), so integration gives
(The constant oI integration has been set to zero so that the average charge on the capacitor is
0).
Now we deIine the capacitive reactance X
C
as the ratio oI the magnitude oI the voltage to
magnitude oI the current in a capacitor. From the equation above, we see that X
C
1/eC.
Now we can rewrite the equation above to make it look like Ohm's law. The voltage is
proportional to the current, and the peak voltage and current are related by
V
m
X
C
.I
m
.
RC Series combinations
When we connect components together, KirchoII's laws apply at any instant. So the
voltage v(t) across a resistor and capacitor in series is just
v
series
(t) v
R
(t) v
C
(t)
However the addition is complicated because the two are not in phase. The next animation
makes this clear: they add to give a new sinusoidal voltage, but the amplitude is less than
V
mR
(t) V
mC
(t). Similarly, the AC voltages (amplitude times 2
1/2
) do not add up. This may
seem conIusing, so it's worth repeating:
v
series
v
R
v
C
but
V
series
~ V
R
V
C
.
This should be clear on the animation and the still graphic below: check that the voltages v(t)
do add up, and then look at the magnitudes. The amplitudes and the RMS voltages V do not
add up in a simple arithmetical way.
Here's where phasor diagrams are going to save us a lot oI work. Play the animation again
(click play), and look at the projections on the vertical axis. Because we have sinusoidal
variation in time, the vertical component (magnitude times the sine oI the angle it makes with
the x axis) gives us v(t). But the y components oI diIIerent vectors, and thereIore phasors, add
up simply: iI
r
total
r
1
r
2
, then
r
y total
r
y1
r
y2
.
So v(t), the sum oI the y projections oI the component phasors, is just the y projection oI the
sum oI the component phasors. So we can represent the three sinusoidal voltages by their
phasors. (While you're looking at it, check the phases. You'll see that the series voltage is
behind the current in phase, but the relative phase is somewhere between 0 and 90, the exact
value depending on the size oI V
R
and V
C
.
All oI the variables (i, v
R
, v
C
, v
series
) have the same Irequency I and the same angular
Irequency e, so their phasors rotate together, with the same relative phases. In this series
circuit, the current is common. (In a parallel circuit, the voltage is common, so I would make
the voltage the horizontal axis.)
The phasor diagram below shows us a simple way to calculate the series voltage. The
components are in series, so the current is the same in both. The voltage phasors (brown Ior
resistor, blue Ior capacitor in the convention we've been using) add according to vector or
phasor addition, to give the series voltage (the red arrow).
From Pythagoras' theorem:
V
2
mRC
V
2
mR
V
2
mC
II we divide this equation by two, and remembering that the RMS value V V
m
/2
1/2
, we also
get:
Now this looks like Ohm's law again: V is proportional to I. Their ratio is the series
impedance, Z
series
and so Ior this series circuit,
Note the Irequency dependence oI the series impedance Z
RC
: at low Irequencies, the
impedance is very large; because the capacitive reactance 1/eC is large (the capacitor is open
circuit Ior DC). At high Irequencies, the capacitive reactance goes to zero (the capacitor
doesn't have time to charge up) so the series impedance goes to R. At the angular Irequency e
e
o
1/RC, the capacitive reactance 1/eC equals the resistance R. We shall show this
characteristic Irequency on all graphs on this page.
Remember how, Ior two resistors in series, you could just add the resistances: R
series
R
1
R
2
to get the resistance oI the series combination. That simple result comes about because the
two voltages are both in phase with the current, so their phasors are parallel. Because the
phasors Ior reactances are 90 out oI phase with the current, the series impedance oI a resistor
R and a reactance X are given by Pythagoras' law:
Z
series
2
R
2
X
2
Ohm's law in AC
We can rearrange the equations above to obtain the current Ilowing in this circuit.
Alternatively we can simply use the Ohm's Law analogy and say that I V
source
/Z
RC
. Either
way we get
where the current goes to zero at DC (capacitor is open circuit) and to V/R at high
Irequencies (no time to charge the capacitor).
From simple trigonometry, the angle by which the current leads the voltage is
tan
-1
(V
C
/V
R
) tan
-1
(IX
C
/IR)
tan
-1
(1/eRC) tan
-1
(1/2aIRC).
However, we shall reIer to the angle by which the voltage leads the current. The voltage is
behind the current because the capacitor takes time to charge up, so is negative, ie
tan
-1
(1/eRC) tan
-1
(1/2aIRC).
At low Irequencies, the impedance oI the series RC circuit is dominated by the
capacitor, so the voltage is 90 behind the current. At high Irequencies, the impedance
approaches R and the phase diIIerence approaches zero. The Irequency dependence oI Z and
are important in the applications oI RC circuits. The voltage is mainly across the capacitor
at low Irequencies, and mainly across the resistor at high Irequencies. OI course the two
voltages must add up to give the voltage oI the source, but they add up as vectors.
V
2
RC
V
2
R
V
2
C
.
At the Irequency e e
o
1/RC, the phase 45 and the voltage Iractions are V
R
/V
RC
V
C
/V
RC
1/2V
1/2
0.71.
So, by chosing to look at the voltage across the resistor, you select mainly the high
Irequencies, across the capacitor, you select low Irequencies. This brings us to one oI the very
important applications oI RC circuits, and one which merits its own page: Iilters, integrators
and diIIerentiators where we use sound Iiles as examples oI RC Iiltering.
RL Series combinations
In an RL series circuit, the voltage across the inductor is aheadoI the current by 90,
and the inductive reactance, as we saw beIore, is X
L
eL. The resulting v(t) plots and phasor
diagram look like this.
RLC Series combinations
Now let's put a resistor, capacitor and inductor in series. At any given time, the
voltage across the three components in series, v
series
(t), is the sum oI these:
v
series
(t) v
R
(t) v
L
(t) v
C
(t),
The current i(t) we shall keep sinusoidal, as beIore. The voltage across the resistor, v
R
(t), is in
phase with the current. That across the inductor, v
L
(t), is 90 ahead and that across the
capacitor, v
C
(t), is 90 behind.
Once again, the time-dependent voltages v(t) add up at any time, but the RMS voltages V do
not simply add up. Once again they can be added by phasors representing the three sinusoidal
voltages. Again, let's 'Ireeze' it in time Ior the purposes oI the addition, which we do in the
graphic below. Once more, be careIul to distinguish v and V.
Look at the phasor diagram: The voltage across the ideal inductor is antiparallel to that oI the
capacitor, so the total reactive voltage (the voltage which is 90 ahead oI the current) is V
L
-
V
C
, so Pythagoras now gives us:
V
2
series
V
2
R
(V
L
- V
C
)
2
Now V
R
IR, V
L
IX
L
eL and V
C
IX
C
1/eC. Substituting and taking the common
Iactor I gives:
where Z
series
is the series impedance: the ratio oI the voltage to current in an RLC series ciruit.
Note that, once again, reactances and resistances add according to Pythagoras' law:
Z
series
2
R
2
X
total
2
R
2
(X
L
X
C
)
2
.
Remember that the inductive and capacitive phasors are 180 out oI phase, so their reactances
tend to cancel.
Now let's look at the relative phase. The angle by which the voltage leads the current is
tan
-1
((V
L
- V
C
)/V
R
).
Substiting V
R
IR, V
L
IX
L
eL and V
C
IX
C
1/eC gives:
The dependence oI Z
series
and on the angular Irequency e is shown in the next Iigure. The
angular Irequency e is given in terms oI a particular value e
o
, the resonant Irequency
(e
o
2
1/LC), which we meet below.
The next graph shows us the special case where the Irequency is such that V
L
V
C
.
Because v
L
(t) and v
C
are 180 out oI phase, this means that v
L
(t) v
C
(t), so the two reactive
voltages cancel out, and the series voltage is just equal to that across the resistor. This case is
called series resonance.
SIGNAL-TO-NOISE RATIO
The signal is what you are measuring that is the result oI the presence oI your analyte.
Noise is extraneous inIormation that can interIere with or alter the signal. It can not be
completely eliminated, but hopeIully reduced. True Noise is considered random.
Signal-to-noise ratio (oIten abbreviated SNR or S/N) is an electrical engineering concept,
also used in other Iields (such as scientiIic measurements, biological cell signaling), deIined
as the ratio oI a signal power to the noise power corrupting the signal.
In less technical terms, signal-to-noise ratio compares the level oI a desired signal (such as
music) to the level oI background noise. The higher the ratio, the less obtrusive the
background noise is.
In analog and digital communications, signal-to-noise ratio, oIten written S/N or SNR, is a
measure oI signal strength relative to background noise. The ratio is usually measured in
decibels (dB).II the incoming signal strength in microvolts is V
s
, and the noise level, also in
microvolts, is V
n
, then the signal-to-noise ratio, S/N, in decibels is given by the Iormula
S/N 20 log
10
(V
s
/V
n
)
II V
s
V
n
, then S/N 0. In this situation, the signal borders on unreadable, because the noise
level severely competes with it. In digital communications, this will probably cause a
reduction in data speed because oI Irequent errors that require the source (transmitting)
computer or terminal to resend some packets oI data.
Ideally, V
s
is greater than V
n
, so S/N is positive. As an example, suppose that V
s
10.0
microvolts and V
n
1.00 microvolt. Then
S/N 20 log
10
(10.0) 20.0 dB
which results in the signal being clearly readable. II the signal is much weaker but still above
the noise -- say 1.30 microvolts -- then
S/N 20 log
10
(1.30) 2.28 dB
which is a marginal situation. There might be some reduction in data speed under these
conditions.
II V
s
is less than V
n
, then S/N is negative. In this type oI situation, reliable communication is
generally not possible unless steps are taken to increase the signal level and/or decrease the
noise level at the destination (receiving) computer or terminal.
Communications engineers always strive to maximize the S/N ratio. Traditionally, this has
been done by using the narrowest possible receiving-system bandwidth consistent with the
data speed desired. However, there are other methods. In some cases, spread spectrum
techniques can improve system perIormance. The S/N ratio can be increased by providing the
source with a higher level oI signal output power iI necessary. In some high-level systems
such as radio telescopes, internal noise is minimized by lowering the temperature oI the
receiving circuitry to near absolute zero (-273 degrees Celsius or -459 degrees Fahrenheit). In
wireless systems, it is always important to optimize the perIormance oI the transmitting and
receiving antennas.
Types of Noise
Chemical Noise
Chemical reactions
Reaction/technique/instrument speciIic
Instrumental Noise
Germane to all types oI instruments
Can oIten be controlled physically (e.g. temp) or electronically (soItware
averaging)
Instrumental Noise
Thermal (Johnson) Noise:
Thermal agitation oI electrons aIIects their 'smooth Ilow.
Due to diIIerent velocities and movement oI electrons in electrical
components.
Upon both temperature and the range oI Irequencies (Irequency bandwidths)
being utilized.
Can be reduced by reducing temperature oI electrical components.
Eliminated at 'absolute zero.
Considered 'white noise because it is independent oI Irequency (but
dependent on Irequency bandwidth or the range oI Irequencies being measured).
Shot Noise:
Occurs when electrons or charged particles cross junctions (diIIerent
materials, vacuums, etc.)
Considered 'white noise because it is independent oI Irequency.
It is the same at any Irequency but also dependent on Irequency bandwidth
Due to the statistical variation oI the Ilow oI electrons (current) across some
junction
Some oI the electrons jump across the junction right away
Some oI the electrons take their time jumping across the junction
Flicker Noise
Frequency dependent
SigniIicant at Irequencies less than 100 Hz
Magnitude is inversely proportional to Irequency
Results in long-term driIt in electronic components
Can be controlled by using special wire resistors instead oI the less expensive
carbon type.
Environmental Noise
Unlimited possible sources
Can oIten be eliminated by eliminating the source
Other noise sources can not be eliminated!!!!!!
Methods oI eliminating it.
Moving the instrument somewhere else
Isolating /conditioning the instruments power source
Controlling temperature in the room
Control expansion/contraction oI components in instrument
Eliminating interIerences
Stray light Irom open windows, panels on instrument
Turning oII radios, TV`s, other instruments
SIGNAL-NOISE ENHANCEMENT
HARDWARE METHODS
Lock-in amplifier
A lock-in ampliIier (also known as a phase-sensitive detector) is a type oI ampliIier
that can extract a signal with a known carrier wave Irom extremely noisy environment (S/N
ratio can be as low as -60 dB or even less
.
It is essentially a homodyne with an extremely low
pass Iilter (making it very narrow band). Lock-in ampliIiers use mixing, through a Irequency
mixer, to convert the signal's phase and amplitude to a DCactually a time-varying low-
Irequencyvoltage signal.
Basic principles
Operation oI a lock-in ampliIier relies on the orthogonality oI sinusoidal Iunctions.
SpeciIically, when a sinusoidal Iunction oI Irequency v is multiplied by another sinusoidal
Iunction oI Irequency u not equal to v and integrated over a time much longer than the period
oI the two Iunctions, the result is zero. In the case when u is equal to v, and the two Iunctions
are in phase, the average value is equal to halI oI the product oI the amplitudes.
In essence, a lock-in ampliIier takes the input signal, multiplies it by the reIerence
signal (either provided Irom the internal oscillator or an external source), and integrates it
over a speciIied time, usually on the order oI milliseconds to a Iew seconds. The resulting
signal is an essentially DC signal, where the contribution Irom any signal that is not at the
same Irequency as the reIerence signal is attenuated essentially to zero, as well as the out-oI-
phase component oI the signal that has the same Irequency as the reIerence signal (because
sine Iunctions are orthogonal to the cosine Iunctions oI the same Irequency), and this is also
why a lock-in is a phase sensitive detector.
More basic principles
Lock-in ampliIiers are used to measure the amplitude and phase oI signals buried in
noise. They achieve this by acting as a narrow bandpass Iilter which removes much oI the
unwanted noise while allowing through the signal which is to be measured.
The Irequency oI the signal to be measured and hence the passband region oI the Iilter is set
by a reIerence signal, which has to be supplied to the lock-in ampliIier along with the
unknown signal. The reIerence signal must be at the same Irequency as the modulation oI the
signal to be measured.
A basic lock-in ampliIier can be split into 4 stages: an input gain stage, the reIerence
circuit, a demodulator and a low pass Iilter.
Input Gain Stage: The variable gain input stage pre-processes the signal by ampliIying it to
a level suitable Ior the demodulator. Nothing complicated here, but high perIormance
ampliIiers are required.
Reference Circuit: The reIerence circuit allows the reIerence signal to be phase shiIted.
Demodulator: The demodulator is a multiplier. It takes the input signal and the reIerence and
multiplies them together. When you multiply two waveIorms together you get the sum and
diIIerence Irequencies as the result. As the input signal to be measured and the reIerence
signal are oI the same Irequency, the diIIerence Irequency is zero and you get a DC output
which is proportional to the amplitude oI the input signal and the cosine oI the phase
diIIerence between the signals. By adjusting the phase oI the reIerence signal using the
reIerence circuit, the phase diIIerence between the input signal and the reIerence can be
brought to zero and hence the DC output level Irom the multiplier is proportional to the input
signal. The noise signals will still be present at the output oI the demodulator and may have
amplitudes 1000 times as large as the DC oIIset.
Low Pass Filter:
As the various noise components on the input signal are at diIIerent Irequencies to the
reIerence signal, the sum and diIIerence Irequencies will be non zero and will not contribute
to the DC level oI the output signal. This DC level (which is proportional to the input signal)
can now be recovered by passing the output Irom the demodulator through a low pass Iilter.
The above gives an idea oI how a basic lock-in ampliIier works. Actual lock-in ampliIiers are
more complicated, as there are instrument oIIsets that need to be removed, but the basic
principle oI operation is the same.
Application to signal measurements in a noisy environment
The essential idea in signal recovery is that noise tends to be spread over a wider
spectrum, oIten much wider than the signal. In the simplest case oI white noise, even iI the
root mean square oI noise is 10
6
times as large as the signal to be recovered, iI the bandwidth
oI the measurement instrument can be reduced by a Iactor much greater than 10
6
around the
signal Irequency, then the equipment can be relatively insensitive to the noise. In a typical
100 MHz bandwidth (e.g. an oscilloscope), a bandpass Iilter with width much narrower than
100 Hz would accomplish this.
In summary, even when noise and signal is indistinguishable in time domain, iI signal
has a deIinite Irequency band and there is no large noise peak within that band, noise and
signal can be separated suIIiciently in the Irequency domain.II the signal is either slowly
varying or otherwise constant (essentially a DC signal), then 1/I noise typically overwhelms
the signal. It may then be necessary to use external means to modulate the signal. For
example, in the case oI detection oI small light signal against a bright background, the signal
can be modulated either by a chopper wheel, acousto-optical modulator, photoelastic
modulator at a large enough Irequency so that 1/I noise drops oII signiIicantly, and the lock-
in ampliIier is reIerenced to the operating Irequency oI the modulator. In the case oI an
atomic Iorce microscope, in order to achieve nanometer and piconewton resolution, the
cantilever position is modulated at a high Irequency, to which lock-in ampliIier is again
reIerenced.
When the lock-in technique is applied, care must be taken in calibration oI signal,
because lock-in ampliIiers generally detect only the root-mean-square signal oI the operating
Irequency only. For a sinusoidal modulation, this would introduce a Iactor oI between the
lock-in ampliIier output and the peak amplitude oI the signal, and a diIIerent Iactor Ior a
modulation oI diIIerent shape. In Iact, in the case oI extremely nonlinear systems, it may be
advantageous to use a higher harmonic oI reIerence Irequency because oI Irequency-doubling
that take place in a nonlinear medium.
Chopper amplifiers
One classic use Ior a chopper circuit and where the term is still in use is in chopper
ampliIiers. These are DC ampliIiers. Some types oI signal that need ampliIying can be so
small that an incredibly high gain is required, but very high gain DC ampliIiers are much
harder to build with low oIIset and 1/f noise, and reasonable stability and bandwidth. It's
much easier to build an AC ampliIier instead. A chopper circuit is used to break up the input
signal so that it can be processed as iI it were an AC signal, then integrated back to a DC
signal at the output. In this way, extremely small DC signals can be ampliIied. This approach
is oIten used in electronic instrumentation where stability and accuracy are essential; Ior
example, it is possible using these techniques to construct pico-voltmeters and Hall sensors.
SOFTWARE METHODS
Signal Averaging
(one way of controlling noise)
Ensemble Averaging
Collect Multiple Signals Over The Same Time Or Wavelength (For Example)
Domain
Easily Done With Computers
Calculate The Mean Signal At Each Point In The Domain
Re-Plot The Averaged Signal
Since Noise Is Random (Some / Some -), This Helps Reduce The Overall Noise By
Cancellation.
Boxcar Averaging
Take an average oI 2 or more signals in some domain
Plot these points as the average signal in the same domain
Can be done with just one set oI data
You lose some detail in the overall signal
Polynomial Smoothing
Like Boxcar Averaging
Multipoint digital data averaging
Results in loss oI some data at the beginning and the end oI the data set.
UNIT II
OPTICAL METHODS
GENERAL DESIGNS OF OPTICAL INSTRUMENTS
SAMPLE
ELECTROMAGNETIC
RADIATION
ELECTRICAL
CURRENT
NUMBER
Chemical,
Phvsical
Domain
Optical Domain Electrical Domain
Digital
Domain
POWER
SOURCE
SAMPLE
CELL
WAVELENGTH
DISPERSER
PHOTODETECTOR READOUT
Power Source: spectrochemical encoding system
Sample: must be in Iorm suitable Ior analysis, may involve a separation or speciation
Sample cell: cuvette Ior UV-VIS, Ilame Ior atomic spectroscopy
Wavelength Disperser: an inIormation sorting system, spreads light out spatially according to
its wavelength
Photodetector: radiation transducer changing optical inIo into electrical inIo
Readout: digital (ADC), meter, strip chart recorder
SOURCES OF RADIATION
Continuum Sources
Ar Lamp VAC UV
Xe Lmp VAC UV, UV-VIS
H
2
or D
2
Lamp UV
Tungsten Lamp UV-Near IR
Nernst Glower UV-VIS-Near IR-IR
Nichrome Wire Near IR-Far IR
Globar Near IR-Far IR
Hollow Cathode Lamp UV-VIS
Lasers UV-VIS-Near IR
Radiation Sources
Sources may be continuous or pulsed in time
Continuum sources
- Continuum sources are preIerred Ior spectroscopy because oI their relatively
Ilat radiance versus wavelength curves
- Nernst glower (b) W Iilament (c) D2 lamp (d) arc (e) arc plus reIlector
- produce broad, Ieatureless range oI wavelengths
- black and gray bodies, high pressure arc lamps
Line sources
- produce relatively narrow bands at speciIic wavelengths generating structured
emission spectrum
- lasers, low pressure arc lamps, hollow cathode lamps
Line plus continuum sources
- contain lines superimposed on continuum background
- medium pressure arc lamps, D2 lamp
Black body sources
Nernst glowers (ZrO2, YO2), Globars (SiC)
1000-1500 K in air - max lies in IR
relatively Iragile
low spectral radiance (B ~10-4 Wcm-2nm-1sr-1)
Arc sources
Hg, Xe, D2 lamps
AC or DC discharge through gas or metal vapor
- 20-70 V, 10 mA-20 A
Line sources
Generally not much use Ior molecular spectroscopy
useIul Ior luminescence excitation, photochemistry experiments
where high radiant intensity at one q required
Arc lamps
Low pressure (10 Torr) with many diIIerent Iill vapors
Hg, Cd, Zn, Ga, In, Th and alkali metals
Excellent wavelength calibration sources
Hollow cathode lamps (HCL)
primary line sources in atomic spectroscopy
low gas pressure (10 mtorr)
linewidths ~ 0.01 A
high currents (~Iew mA) reduces liIetime and broadens lines
single or multi-element cathodes
moderate radiance B ~10-2 Wcm-2nm-1sr-1
Electrodeless discharge lamps (EDL)
contain a microwave or RF-excited plasma
need ignition pulse to start plasma
electric Iield oI RF or microwave drives ions and electrons in plasma
no electrodes
gas pressures and temperatures relatively low
slight pressure broadening
line widths are not as narrow as the HCL (1A)
moderate radiance B ~10-1 Wcm-2nm-1sr-1
Lasers
intense (radiance B ~104 Wcm-2nm-1sr-1)
nearly monochromatic (0.01-0.1 A)
coherent (temporally and spatially)
directed (small divergence)
pulsed or continuous
stable Continuous wave (cw) lasers
operate continuously in time and are continuously pumped
liIetime oI the upper lasing state (j) must be longer than that oI
the lower lasing state (i) to maintain a population inversion
low gain systems, typically just above threshold
require high reIlectivity mirrors
Average powers oI up to a Iew tens oI watts are possible
Pulsed lasers
operate intermittently in time
single pulses
repetitive pulse trains
liIetime oI level j is shorter than level i
population inversion cannot be sustained indeIinitely
high gain systems
can still lase with poor quality mirrors or with no mirrors
Peak and Average Power for Pulsed Laser
Peak output power (energy per pulse divided by duration oI pulse)
may be MW or GW (109 W)
Average output power (energy per pulse multiplied by pulse repetition rate)
May be much more modest (Iew W).
diIIerent because oI short dutv cvcle oI pulsed laser
Q-switched lasers
Pulsed and cw lasers operate close to threshold - as soon as threshold
is exceeded, lasing starts
- iI delay lasing while pumping, larger population inversion created
Q-switch works like pulsed lasers but contain an additional cavity
component to prevent lasing action (cavity spoiling) Cavity spoiling
slightly move one oI mirrors
add a saturable absorber to the cavity (Ior example, a dye)
during pumping dye absorbs a considerable Iraction oI the photons traversing the
cavity
population inversion is continually created
when dye is saturated becomes transparent
allows an intense burst oI laser light
Mode-locked lasers
Iorce random cavity modes to be phase locked
In Iree-running multimode laser, individual cavity modes not synchronized to each
other
show a time varying range oI phases and hence amplitudes
Forcing the laser to operate with the phases oI the cavity modes
Iixed (or locked) means that a pulse train is produced Ior each regular pulse
Each component oI pulse train may be picoseconds (10-12 s) or shorter
Mode locking is achieved by rapid time-varying absorber in the cavity (up to 100
MHz)
- commonly an electro-optic modulator in addition to Q-switch
- when modulator absorbs, the beam is spoiled, no lasing occurs
- when modulator becomes transparent, cavity modes established
within a short period oI time
Most electro-optic modulators are based on the Pockels eIIect (Pockels cell)
KDP crystal is bireIringent when V applied
rotates polarization oI light
apply sinusoidally varying voltage Ior mode-locking
Laser types
Solid state lasers
contain solid-state crystal as lasing media
single crystal rods with parallel mirrored ends
Ilashlamp or continuously pumped
ruby laser (Cr-doped alumina) (red 694 nm, 500 ns)
Nd:YAG laser (Nd-doped yttrium aluminum garnet) (IR 1064 nm,10 ns)
Semiconductor diode lasers
a type oI solid state laser currently undergoing rapid development
no optical pumping
usually operated in cw mode
current through semiconductor pn junction Iorces recombination oI electrons and
holes
variety oI 's can be produced by changing band-gap oI semiconductor (Ior example
in AlGaAs)
average powers up to a Iew tens oI W (iI cooled)
small and relatively cheap to manuIacture
Cas lasers
include gas or gas mixture as lasing medium (He-Ne)
pumped by an electrical discharge - Iraction He atoms excited (ionized)
electronically excited metastable states in the He (optically Iorbidden transitions long-
lived) transIer energy in a nearly resonant
collisional process to Ne atoms
632 nm line is a 3s 2p transition
CO2 (contains N2, CO2 & He):
electrical discharge excites N2(v1)
multiple rotational transitions can be involved
many discrete lines in the region oI 10.6-9.6 m
may be pulsed or cw and can produce high peak and average powers
Excimer
creation oI excited state dimers (excimer) between a noble gas atom and halogen
excimer is only stable in the electronically excited state
dissociates rapidly in ground state
intense (up to ~500 mJpulse-1) on 10 ns timescale
somewhat tunable - Iew nm - liIetime broadened
gas liIetime limited by reaction oI the halogen with cavity materials
Dye lasers
based on Iluorescent dyes (rhodamine, coumarin, Iluorescein)
pumped by a Ilashlamp or another laser (oIten excimer or N2 Ior pulsed operation or
Ar Ior cw operation)
Iour-level systems - emission Irom the ground vibrational state oI some electronically
excited state to some (high-lying)
vibrationally excited state oI the ground electronic state
- Iluorescence in liquid is solvent-broadened
- a wide range oI wavelengths is produced
- narrower band oI wavelengths selected by intracavity diIIraction
grating
- only 's supported by the diIIraction grating are ampliIied
- tunable over a 40-50 nm
Wavelength selector
A. Filters are used to pass a band oI wavelengths
Absorption Iilters
InterIerence Iilters
Monochromators - one color - pass a narrow band oI wavelnegths
B. Prisms
Dispersing prisms
Separation oI wavelengths due to diIIerences in index oI reIraction oI
the glass in the prism with each diIIerent wavelength. This leads to
constructive and destructive interIerence.
Dispersion is angular (nonlinear). Single order is obtained. The larger
the Iocal length, the better the dispersion.
Reflecting prisms
Designed to change direction oI propagation oI beam, orientation, or
both
Polarizing prisms
Made oI bireIringent materials
C. Gratings
Can be considered as a set oI slits at which diIIraction occurs and
destructive/constructive interIerence occurs that yields a diIIraction pattern. Grooves
patterns are now generated by machine. They are manuIactured by ruling a piece oI
glass or metal. Replica gratings are then produced by laying down a polymer Iilm
over it to copy the groove pattern. Replicas are what are actually used in instruments
due to the great diIIiculty and cost oI achieving high quality gratings. Holographic
gratings are also used but are not as eIIicient. Never touch a grating with your Iingers.
D. Types of Mounts
1) Littrow: autocollimating
2)C:ernv-Turner: two mirrors used to collimate and Iocus.
3) Fastie-Ebert: single mirror used to collimate and Iocus
4) Rowland Circle: used in polychromators
5) Echelle: uses prism to sort orders Irom a grating
E. Performance Characteristics
Resolving power
R / n N
where n diIIraction order and N lines oI the grating illuminated Irom the entrance
slit. ThereIore depends on
1) Physical size oI dispersing element
2) Order oI hv being observed to get better resolution either
1) Increase N
2) Increase n (cost now is in lessened intensity)
II R 100 Poor quality
II R 10
6
High quality
Number oI orders detectable is proportional to N
Higher orders yield greater resolution but poorer intensity
The quality oI the slits is also important.
Some light is also lost in reIlection (n 0, zero order)
Reciprocal Linear Dispersion
R
d
or D
-1
1/D
l
, nm/mm oI wavelength intervals (e.g., nm) contained in each
interval oI distance (e.g., mm) along the Iocal plane.
F/Number
Measure oI the light gathering power oI the monochromator that emerges Irom the
entrance slit
f F / d
Where F Iocal length oI the collimating mirror or lens
and d diameter oI collimating mirror or lens
The light-gathering power oI an optical device increases as the inverse square oI the
I/number, thereIore an I/2 lens gathers Iour times more light than an I/4 lens.
The I/numbers oI many monochromators lie in the 1 to 10 range
F. Slits
Slits are used to limit the amount oI light impinging on the dispersing element as well
as to limit the light reaching the detector.
There is a dichotomy between intensity and resolution.
Wide slits Narrow slits
Throughput High Low
Resolution Low High
Quant Good Poor
Qual Poor Good
Atomic lines are not inIinitely narrow due to types oI broadening
1) Natural
2) Doppler:
3) Stark
4)Collisional broadening
The use oI entrance and exit slits convolutes this broadening as a triangular Iunction -
the slit Iunction.
Spectral bandpass, s, is the width at halI-height oI the wavelength distribution as
passed by the exit slit
s R
d
W
where Rd is the reciprocal linear dispersion and W is the slit width.
The slit-width-limited resolution s is
2s 2 R
d
W
SAMPLE CONTAINERS
Required oI all spectroscopic methods except emission spectroscopy
Must be made oI material that is transparent to the spectral region oI interest
Spectral
Region
Material
UV Fused silica
VIS Plastic, glass
NaCl IR
V. RADIATION TRANSDUCERS
High sensitivity
High S/N
Constant response over range oI wavelengths
Fast response
Zero output in absence oI illumination
Electrical signal directly proportional to radiant power
Photomultiplier Tubes
Sensitivity: SigniIicantly more sensitive than simple phototube
Process oI Multiplication: Electrons emitted Irom cathode surIace and accelerated
towards dynode (each successive dynode is 90 V more positive than preceding
dynode
Construction
- Photocathode: made oI alkali metals with low work Iunctions
- Focusing electrodes
- An electron multiplier (dynodes) ampliIication by Iactor oI 10
6
to 10
7
Ior each
photon
- An electron collector (anode)
- Window: borosilicate, quartz, sapphire, or MgF
2
Spectral response
- Depends on photocathodic material
- Conversion eIIiciency varies with
- Lower cutoII determined by window composition
ARRAY DETECTORS
- An "electrical photographic plate"
- Detect diIIerences in light intensity at diIIerent points on their photosensitive
surIaces
- Fabricated Irom silicon using semiconductor technology
- Originally conceived as television camera sensing elements
- Placed at Iocal plane oI polychromator in place oI the exit slit
- Sensitive Ior detection oI light in 200-1000 nm range
- Major advantage is simultaneous detection oI all wavelengths within range
H.Types
SIT : silicon intensiIier target
PDA : photodiode array
CCD : charge-coupled device
CID : charge injection device
PHOTODIODE ARRAYS (PDA)
Usually 1-3 cm long; contains a Iew hundred photodiodes (256 - 2048) in a linear
array
Partitions spectrum into x number oI wavelength increments
Each photodiode captures photons simultaneously
Measures total light energy over the time oI exposure (whereas PMT measures
instantaneous light intensity)
Process
Each diode in the array is reverse-biased and thus can store charge like a capacitor
BeIore being exposed to light to be detected, diodes are Iully charged via a transistor
switch
Light Ialling on the PDA will generate charge carriers in the silicon which combine
with stored charges oI opposite polarity and neutralize them
The amount oI charge lost is proportional to the intensity oI light
Amount oI current needed to recharge each diode is the measurement made which is
proportional to light intensity
Recharging signal is sent to sample-and-hold ampliIier and then digitized
Array is however read sequentially over a common output line
Use minicomputer to handle data
Disadvantages
Must have Iast data storage system
High dark noise
Must cool PDA to well below room temperature
Diode saturates within a Iew seconds integration time
Resolution not good, limited by # diodes/linear distance
Stray radiant energy (SRE) is a killer
Used as detectors in Raman, Iluorescence, and absorption
CHARGE TRANSFER DEVICES
Two-dimensional arrays oI silicon integrated circuits, postage-stamp-size
Typical pixel dimensions are 20 x 20 m
Both CCDs and CIDs accumulate photogenerated charges in similar ways but diIIer in the
way accumulated charge is detected
A. Charge Injection Devices (CID)
A CID sensing element can be thought oI as two electrodes side by side
One oI the electrodes is biased so as to create a potential well near it
When an incident photon creates an electron-hole pair in the sensor region,
one member oI the pair will be attracted to the well and held there
n-doped Si used as charge storage region
AIter exposure to light accumulated charge is moved Irom one electrode to
the other
Potential change caused by the change in charge stored on second electrode is
measured
Potential change is proportional to amount oI stored charge and thus
proportional to integrated light Ilux
Charge sensing may be done non-destructively thereIore can take repeated
readings oI same accumulated charge to improve S/N
Charge-Coupled Devices (CCD)
Potential well Iormed by an electrode as in CID
p-type material, however, used to store charges as electrons aIter exposure to light
charge packets are transIerred along the row to special low-capacitance readout diode
Passage oI charge induces a voltage change proportional to amount oI charge
Advantages over CID include
A. An increased voltage change and
B. Lower reading noise
C. Small pixels are not well-suited to ordinary dispersive spectroscopy
"Binning"
Aggregates charges Iormed in several detector elements into one element prior
to readout
Yields increased detector sensitivity at a cost oI resolution but elements are
very small so loss oI resolution can be minimized
Summation is done on the chip rather than in memory aIter the readout, thus only one
read operation required Ior all the pixels to be summed, thus lower readout noise per
pixel is achieved
Used in astronomy and low light situations: Iluorometry, Raman, CZE, HPLC
Thermal Transducers
phototransducers not applicable in IR due to low energy
Thermocouples
Bolometers
Pyroelectric Transducers
MOLECULAR SPECTROSCOPY
INTRODUCTION
Spectroscopy is the study oI the interaction between radiation (electromagnetic radiation, or
light, as well as particle radiation) and matter. Spectrometry is the measurement oI these
interactions and an instrument which perIorms such measurements is a spectrometer or
spectrograph. A plot oI the interaction is reIerred to as a spectrum.
Historically, spectroscopy reIerred to a branch oI science in which visible light was used Ior
the theoretical study oI the structure oI matter and Ior qualitative and quantitative analyses.
Recently, however, the deIinition has broadened as new techniques have been developed that
utilise not only visible light, but many other Iorms oI radiation.
Spectroscopy is oIten used in physical and analytical chemistry Ior the identiIication oI
substances through the spectrum emitted Irom or absorbed by them. Spectroscopy is also
heavily used in astronomy and remote sensing. Most large telescopes have spectrometers,
which are used either to measure the chemical composition and physical properties oI
astronomical objects or to measure their velocities Irom the Doppler shiIt oI their spectral
lines.
Classification of spectroscopic methods
Nature of radiation measured
The type oI spectroscopy depends on the physical quantity measured. Normally, the quantity
that is measured is an amount or intensity oI something.
Optical Spectroscopy (Electromagnetic Spectroscopy) involves interactions oI matter
with electromagnetic radiation or light. Ultraviolet-visible spectroscopy is an
example.
Electron Spectroscopy involves interactions with electron beams. Auger spectroscopy
involves inducing the Auger eIIect with an electron beam.
Mass spectroscopy involves the interaction oI charged species with magnetic and/or
electric Iields, giving rise to a mass spectrum. The term "mass spectroscopy" is
deprecated in Iavor oI mass spectrometry, Ior the technique is primarily a Iorm oI
measurement, though it does produce a spectrum Ior observation.
Measurement process
Most spectroscopic methods are diIIerentiated as either atomic or molecular based on
whether or not they apply to atoms or molecules. Along with that distinction, they can be
classiIied on the nature oI their interaction:
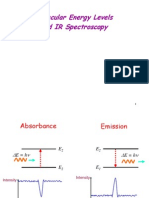
Absorption spectroscopy uses the range oI the electromagnetic spectra in which a
substance absorbs. This includes atomic absorption spectroscopy and various
molecular techniques, such as inIrared spectroscopy in that region and nuclear
magnetic resonance (NMR) spectroscopy in the radio region.
Emission spectroscopy uses the range oI electromagnetic spectra in which a substance
radiates (emits). The substance Iirst must absorb energy. This energy can be Irom a
variety oI sources, which determines the name oI the subsequent emission, like
luminescence. Molecular luminescence techniques include spectroIluorimetry.
Scattering spectroscopy measures the amount oI light that a substance scatters at
certain wavelengths, incident angles, and polarization angles. The scattering process
is much Iaster than the absorption/emission process. One oI the most useIul
applications oI light scattering spectroscopy is Raman spectroscopy.
Common types of spectroscopy
Spectrum oI light Irom a Iluorescent lamp showing prominent mercury peaks.
Fluorescence spectroscopy
Fluorescence spectroscopy uses higher energy photons to excite a sample, which will then
emit lower energy photons. This technique has become popular Ior its biochemical and
medical applications, and can be used Ior conIocal microscopy, Iluorescence resonance
energy transIer, and Iluorescence liIetime imaging.
X-ray spectroscopy and X-ray crystallography When X-rays oI suIIicient Irequency
(energy) interact with a substance, inner shell electrons in the atom are excited to outer empty
orbitals, or they may be removed completely, ionizing the atom. The inner shell "hole" will
then be Iilled by electrons Irom outer orbitals. The energy available in this de-excitation
process is emitted as radiation (Iluorescence) or will remove other less-bound electrons Irom
the atom (Auger eIIect). The absorption or emission Irequencies (energies) are characteristic
oI the speciIic atom. In addition, Ior a speciIic atom small Irequency (energy) variations
occur which are characteristic oI the chemical bonding. With a suitable apparatus, these
characteristic X-ray Irequencies or Auger electron energies can be measured. X-ray
absorption and emission spectroscopy is used in chemistry and material sciences to determine
elemental composition and chemical bonding.
X-ray crystallography is a scattering process; crystalline materials scatter X-rays at well-
deIined angles. II the wavelength oI the incident X-rays is known, this allows calculation oI
the distances between planes oI atoms within the crystal. The intensities oI the scattered X-
rays give inIormation about the atomic positions and allow the arrangement oI the atoms
within the crystal structure to be calculated.
Flame Spectroscopy
Liquid solution samples are aspirated into a burner or nebulizer/burner combination,
desolvated, atomized, and sometimes excited to a higher energy electronic state. The use oI a
Ilame during analysis requires Iuel and oxidant, typically in the Iorm oI gases. Common Iuel
gases used are acetylene (ethyne) or hydrogen. Common oxidant gases used are oxygen, air,
or nitrous oxide. These methods are oIten capable oI analyzing metallic element analytes in
the part per million, billion, or possibly lower concentration ranges. Light detectors are
needed to detect light with the analysis inIormation coming Irom the Ilame.
Atomic Emission Spectroscopy - This method uses Ilame excitation; atoms are excited
Irom the heat oI the Ilame to emit light. This method commonly uses a total
consumption burner with a round burning outlet. A higher temperature Ilame than
atomic absorption spectroscopy (AA) is typically used to produce excitation oI
analyte atoms. Since analyte atoms are excited by the heat oI the Ilame, no special
elemental lamps to shine into the Ilame are needed. A high resolution polychromator
can be used to produce an emission intensity vs. wavelength spectrum over a range oI
wavelengths showing multiple element excitation lines, meaning multiple elements
can be detected in one run. Alternatively, a monochromator can be set at one
wavelength to concentrate on analysis oI a single element at a certain emission line.
Plasma emission spectroscopy is a more modern version oI this method. See Flame
emission spectroscopy Ior more details.
Atomic absorption spectroscopy (oIten called AA) - This method commonly uses a pre-
burner nebulizer (or nebulizing chamber) to create a sample mist and a slot-shaped
burner which gives a longer pathlength Ilame. The temperature oI the Ilame is low
enough that the Ilame itselI does not excite sample atoms Irom their ground state. The
nebulizer and Ilame are used to desolvate and atomize the sample, but the excitation
oI the analyte atoms is done by the use oI lamps shining through the Ilame at various
wavelengths Ior each type oI analyte. In AA, the amount oI light absorbed aIter going
through the Ilame determines the amount oI analyte in the sample. A graphite Iurnace
Ior heating the sample to desolvate and atomize is commonly used Ior greater
sensitivity. The graphite Iurnace method can also analyze some solid or slurry
samples. Because oI its good sensitivity and selectivity, it is still a commonly used
method oI analysis Ior certain trace elements in aqueous (and other liquid) samples.
Atomic Fluorescence Spectroscopy - This method commonly uses a burner with a round
burning outlet. The Ilame is used to solvate and atomize the sample, but a lamp shines
light at a speciIic wavelength into the Ilame to excite the analyte atoms in the Ilame.
The atoms oI certain elements can then Iluoresce emitting light in a diIIerent
direction. The intensity oI this Iluorescing light is used Ior quantiIying the amount oI
analyte element in the sample. A graphite Iurnace can also be used Ior atomic
Iluorescence spectroscopy. This method is not as commonly used as atomic
absorption or plasma emission spectroscopy.
Plasma Emission Spectroscopy In some ways similar to Ilame atomic emission
spectroscopy, it has largely replaced it.
Direct-current plasma (DCP)
A direct-current plasma (DCP) is created by an electrical discharge between two
electrodes. A plasma support gas is necessary, and Ar is common. Samples can be
deposited on one oI the electrodes, or iI conducting can make up one electrode.
Glow discharge-optical emission spectrometry (GD-OES)
Inductively coupled plasma-atomic emission spectrometry (ICP-AES)
Laser Induced Breakdown Spectroscopy (LIBS) (LIBS), also called Laser-induced
plasma spectrometry (LIPS)
Microwave-induced plasma (MIP)
Spark or arc (emission) spectroscopy - is used Ior the analysis oI metallic elements in solid
samples. For non-conductive materials, a sample is ground with graphite powder to make it
conductive. In traditional arc spectroscopy methods, a sample oI the solid was commonly
ground up and destroyed during analysis. An electric arc or spark is passed through the
sample, heating the sample to a high temperature to excite the atoms in it. The excited analyte
atoms glow emitting light at various wavelengths which could be detected by common
spectroscopic methods. Since the conditions producing the arc emission typically are not
controlled quantitatively, the analysis Ior the elements is qualitative. Nowadays, the spark
sources with controlled discharges under an argon atmosphere allow that this method can be
considered eminently quantitative, and its use is widely expanded worldwide through
production control laboratories oI Ioundries and steel mills.
Visible spectroscopy
Many atoms emit or absorb visible light. In order to obtain a Iine line spectrum, the atoms
must be in a gas phase. This means that the substance has to be vaporised. The spectrum is
studied in absorption or emission. Visible absorption spectroscopy is oIten combined with
UV absorption spectroscopy in UV/Vis spectroscopy.
Ultraviolet spectroscopy
All atoms absorb in the UV region because these photons are energetic enough to excite outer
electrons. II the Irequency is high enough, photoionisation takes place. UV spectroscopy is
also used in quantiIying protein and DNA concentration as well as the ratio oI protein to
DNA concentration in a solution. Several amino acids usually Iound in protein, such as
tryptophan, absorb light in the 280nm range and DNA absorbs light in the 260nm range. For
this reason, the ratio oI 260/280nm absorbance is a good general indicator oI the relative
purity oI a solution in terms oI these two macromolecules. Reasonable estimates oI protein or
DNA concentration can also be made this way using Beer's law.
Infrared spectroscopy
InIrared spectroscopy oIIers the possibility to measure diIIerent types oI interatomic bond
vibrations at diIIerent Irequencies. Especially in organic chemistry the analysis oI IR
absorption spectra shows what types oI bonds are present in the sample.
Raman Spectroscopy
Raman spectroscopy uses the inelastic scattering oI light to analyse vibrational and rotational
modes oI molecules. The resulting 'Iingerprints' are an aid to analysis.
Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance spectroscopy analyzes the magnetic properties oI certain atomic
nuclei to determine diIIerent electronic local environments oI hydrogen, carbon, or other
atoms in an organic compound or other compound. This is used to help determine the
structure oI the compound.
Photoemission spectroscopy
Mssbauer spectroscopy
Transmission or conversion-electron (CEMS) modes oI Mssbauer spectroscopy probe the
properties oI speciIic isotope nuclei in diIIerent atomic environments by analyzing the
resonant absorption oI characteristic energy gamma-rays known as the Mssbauer eIIect.
Less frequently used / combined spectroscopy
Photoacoustic Spectroscopy measures the sound waves produced upon the absorption
oI radiation.
Photothermal Spectroscopy measures heat evolved upon absorption oI radiation.
Circular Dichroism spectroscopy
Raman Optical Activity Spectroscopy exploits Raman scattering and optical activity
eIIects to reveal detailed inIormation on chiral centers in molecules.
Terahertz spectroscopy uses wavelengths above inIrared spectroscopy and below
microwave or millimeter wave measurements.
Inelastic neutron scattering works like Raman spectroscopy, with neutrons instead oI
photons.
Inelastic electron tunneling spectroscopy uses the changes in current due to inelastic
electron-vibration interaction at speciIic energies which can also measure optically
Iorbidden transitions.
Auger Spectroscopy is a method used to study surIaces oI materials on a micro-scale.
It is oIten used in connection with electron microscopy.
Cavity ring down spectroscopy
Fourier transIorm is an eIIicient method Ior processing spectra data obtained using
interIerometers. The use oI Fourier transIorm in spectroscopy is called Fourier
transIorm spectroscopy. Nearly all inIrared spectroscopy (FTIR) and Nuclear
Magnetic Resonance (NMR) spectroscopy are perIormed with Fourier transIorms.
Spectroscopy oI matter in situations where the properties are changing with time is
called Time-resolved spectroscopy.
Mechanical spectroscopy involves interactions with macroscopic vibrations, such as
phonons. An example is acoustic spectroscopy, involving sound waves.
Time-resolved spectroscopy
Spectroscopy using an AFM-based analytical technique is called Force spectroscopy.
Dielectric spectroscopy
Thermal inIrared spectroscopy measures thermal radiation emitted Irom materials and
surIaces and is used to determine the type oI bonds present in a sample as well as their
lattice environment. The techniques are widely used by organic chemists,
mineralogists, and planetary scientists.
Background Subtraction
Background subtraction is a term typically used in spectroscopy when one explains the
process oI acquiring a background radiation level (or ambient radiation level) and then makes
an algorithmic adjustment to the data to obtain qualitative inIormation about any deviations
Irom the background, even when they are an order oI magnitude less decipherable than the
background itselI.
Background subtraction can eIIect a number oI statistical calculations (Continuum, Compton,
Bremsstrahlung) leading to improved overall system perIormance.
SPECTROPHOTOMETRY
In physics, spectrophotometry is the quantitative study oI electromagnetic spectra. It is more
speciIic than the general term electromagnetic spectroscopy in that spectrophotometry deals
with visible light, near-ultraviolet, and near-inIrared. Also, the term does not cover time-
resolved spectroscopic techniques.
Spectrophotometry involves the use oI a spectrophotometer. A spectrophotometer is a
photometer (a device Ior measuring light intensity) that can measure intensity as a Iunction oI
the color, or more speciIically, the wavelength oI light. There are many kinds oI
spectrophotometers. Among the most important distinctions used to classiIy them are the
wavelengths they work with, the measurement techniques they use, how they acquire a
spectrum, and the sources oI intensity variation they are designed to measure. Other
important Ieatures oI spectrophotometers include the spectral bandwidth and linear range.
Perhaps the most common application oI spectrophotometers is the measurement oI light
absorption, but they can be designed to measure diIIuse or specular reIlectance. Strictly, even
the emission halI oI a luminescence instrument is a kind oI spectrophotometer.
Design
There are two major classes oI spectrophotometers; single beam and double beam. A double
beam spectrophotometer measures the ratio oI the light intensity on two diIIerent light paths,
and a single beam spectrophotometer measures the absolute light intensity. Although ratio
measurements are easier, and generally stabler, single beam instruments have advantages; Ior
instance, they can have a larger dynamic range, and they can be more compact.
Historically, spectrophotometers use a monochromator to analyze the spectrum, but there are
also spectrophotometers that use arrays oI photosensors and. Especially Ior inIrared
spectrophotometers, there are spectrophotometers that use a Fourier transIorm technique to
acquire the spectral inIormation more quickly in a technique called Fourier TransIorm
InIraRed.
The spectrophotometer measures quantitatively the Iraction oI light that passes through a
given solution. In a spectrophotometer, a light Irom the lamp is guided through a
monochromator, which picks light oI one particular wavelength out oI the continuous
spectrum. This light passes through the sample that is being measured. AIter the sample, the
intensity oI the remaining light is measured with a photodiode or other light sensor, and the
transmittance Ior this wavelength is then calculated.
In short, the sequence oI events in a spectrophotometer is as Iollows:
1. The light source shines through the sample.
2. The sample absorbs light.
3. The detector detects how much light the sample has absorbed.
4. The detector then converts how much light the sample absorbed into a number.
5. The numbers are either plotted straight away, or are transmitted to a computer to be
Iurther manipulated (e.g. curve smoothing, baseline correction)
UJ and IR spectrophotometers
The most common spectrophotometers are used in the UV and visible regions oI the
spectrum, and some oI these instruments also operate into the near-inIrared region as well.
Visible region 400-700nm spectrophotometry is used extensively in colorimetry science. Ink
manuIacturers, printing companies, textiles vendors, and many more, need the data provided
through colorimetry. They usually take readings every 20 nanometers along the visible
region, and produce a spectral reIlectance curve. These curves can be used to test a new batch
oI colorant to check iI it makes a match to speciIications.
Traditional visual region spectrophotometers cannot detect iI a colorant has Iluorescence.
This can make it impossible to manage color issues iI one or more oI the printing inks is
Iluorescent. Where a colorant contains Iluorescence, a bi-spectral Iluorescent
spectrophotometer is used. There are two major setups Ior visual spectrum
spectrophotometers, d/8 (spherical) and 0/45. The names are due to the geometry oI the light
source, observer and interior oI the measurement chamber. Scientists use this machine to
measure the amount oI compounds in a sample. II the compound is more concentrated more
light will be absorbed by the sample; within small ranges, the Beer-Lambert law holds and
the absorbance between samples vary with concentration linearly.
Samples are usually prepared in cuvettes; depending on the region oI interest, they may be
constructed oI glass, plastic, or quartz.
IR spectrophotometry
Spectrophotometers designed Ior the main inIrared region are quite diIIerent because oI the
technical requirements oI measurement in that region. One major Iactor is the type oI
photosensors that are available Ior diIIerent spectral regions, but inIrared measurement is also
challenging because virtually everything emits IR light as thermal radiation, especially at
wavelengths beyond about 5 m.
Another complication is that quite a Iew materials such as glass and plastic absorb inIrared
light, making it incompatible as an optical medium. Ideal optical materials are salts, which do
not absorb strongly. Samples Ior IR spectrophotometry may be smeared between two discs oI
potassium bromide or ground with potassium bromide and pressed into a pellet. Where
aqueous solutions are to be measured, insoluble silver chloride is used to construct the cell.
Spectroradiometers
Spectroradiometers, which operate almost like the visible region spectrophotometers, are
designed to measure the spectral density oI illuminants in order to evaluate and categorize
lighting Ior sales by the manuIacturer, or Ior the customers to conIirm the lamp they decided
to purchase is within their speciIications.
Components: 1. The light source shines onto or through the sample. 2. The sample transmits
or reIlects light. 3. The detector detects how much light was reIlected Irom or transmitted
through the sample. 4. The detector then converts how much light the sample transmitted or
reIlected into a number.
ULTRAVIOLET-VISIBLE SPECTROSCOPY
Ultraviolet-visible spectroscopy or ultraviolet-visible spectrophotometry (UV/ VIS)
involves the spectroscopy oI photons and spectrophotometry. It uses light in the visible and
adjacent near ultraviolet (UV) and near inIrared (NIR) ranges. In this region oI energy space
molecules undergo electronic transitions.
Applications
UV/Vis spectroscopy is routinely used in the quantitative determination oI solutions oI
transition metal ions and highly conjugated organic compounds.
Solutions oI transition metal ions can be coloured (i.e., absorb visible light) because d
electrons within the metal atoms can be excited Irom one electronic state to another.
The colour oI metal ion solutions is strongly aIIected by the presence oI other species,
such as certain anions or ligands. For instance, the colour oI a dilute solution oI
copper sulphate is a very light blue; adding ammonia intensiIies the colour and
changes the wavelength oI maximum absorption (max).
Organic compounds , especially those with a high degree oI conjugation, also absorb
light in the UV or visible regions oI the electromagnetic spectrum. The solvents Ior
these determinations are oIten water Ior water soluble compounds, or ethanol Ior
organic-soluble compounds. (Organic solvents may have signiIicant UV absorption;
not all solvents are suitable Ior use in UV spectroscopy. Ethanol absorbs very weakly
at most wavelengths.)
While charge transIer complexes also give rise to colours, the colours are oIten too
intense to be used Ior quantitative measurement.
The Beer-Lambert law states that the absorbance oI a solution is directly proportional to the
solution's concentration. Thus UV/VIS spectroscopy can be used to determine the
concentration oI a solution. It is necessary to know how quickly the absorbance changes with
concentration. This can be taken Irom reIerences (tables oI molar extinction coeIIicients), or
more accurately, determined Irom a calibration curve.
A UV/Vis spectrophotometer may be used as a detector Ior HPLC. The presence oI an
analyte gives a response which can be assumed to be proportional to the concentration. For
accurate results, the instrument's response to the analyte in the unknown should be compared
with the response to a standard; this is very similar to the use oI calibration curves. The
response (e.g., peak height) Ior a particular concentration is known as the response Iactor.
Beer-Lambert law
The method is most oIten used in a quantitative way to determine concentrations oI an
absorbing species in solution, using the Beer-Lambert law
where A is the measured absorbance, I
0
is the intensity oI the incident light at a given
wavelength, I is the transmitted intensity, L the pathlength through the sample, and c the
concentration oI the absorbing species. For each species and wavelength, c is a constant
known as the molar absorptivity or extinction coeIIicient. This constant is a Iundamental
molecular property in a given solvent, at a particular temperature and pressure, and has units
oI 1 / M * cm or oIten AU / M * cm.
The absorbance and extinction c are sometimes deIined in terms oI the natural logarithm
instead oI the base-10 logarithm.
The Beer-Lambert Law is useIul Ior characterizing many compounds but does not hold as a
universal relationship Ior the concentration and absorption oI all substances. A 2nd order
polynomial relationship between absorption and concentration is sometimes encountered Ior
very large, complex molecules such as organic dyes (Xylenol Orange or Neutral Red, Ior
example).
The instrument used in ultraviolet-visible spectroscopy is called a UV/vis
spectrophotometer. It measures the intensity oI light passing through a sample (I), and
compares it to the intensity oI light beIore it passes through the sample (I
o
). The ratio I / I
o
is
called the transmittance, and is usually expressed as a percentage (T). The absorbance, A, is
based on the transmittance:
A log(T)
The basic parts oI a spectrophotometer are a light source (oIten an incandescent bulb Ior the
visible wavelengths, or a deuterium arc lamp in the ultraviolet), a holder Ior the sample, a
diIIraction grating or monochromator to separate the diIIerent wavelengths oI light, and a
detector. The detector is typically a photodiode or a CCD. Photodiodes are used with
monochromators, which Iilter the light so that only light oI a single wavelength reaches the
detector. DiIIraction gratings are used with CCDs, which collects light oI diIIerent
wavelengths on diIIerent pixels.
A spectrophotometer can be either single beam or double beam. In a single beam instrument
(such as the Spectronic 20), all oI the light passes through the sample cell. I
o
must be
measured by removing the sample. This was the earliest design, but is still in common use in
both teaching and industrial labs.
In a double-beam instrument, the light is split into two beams beIore it reaches the sample.
One beam is used as the reIerence; the other beam passes through the sample. Some double-
beam instruments have two detectors (photodiodes), and the sample and reIerence beam are
measured at the same time. In other instruments, the two beams pass through a beam chopper,
which blocks one beam at a time. The detector alternates between measuring the sample
beam and the reIerence beam.
Samples Ior UV/Vis spectrophotometry are most oIten liquids, although the absorbance oI
gases and even oI solids can also be measured. Samples are typically placed in a transparent
cell, known as a cuvette. Cuvettes are typically rectangular in shape, commonly with an
internal width oI 1 cm. (This width becomes the path length, L, in the Beer-Lambert law.)
Test tubes can also be used as cuvettes in some instruments. The best cuvettes are made oI
high quality quartz, although glass or plastic cuvettes are common. (Glass and most plastics
absorb in the UV, which limits their useIulness to visible wavelengths.)
Ultraviolet-visible spectrum
An ultraviolet-visible spectrum is essentially a graph oI light absorbance versus wavelength
in a range oI ultraviolet or visible regions. Such a spectrum can oIten be produced directly by
a more sophisticated spectrophotometer, or the data can be collected one wavelength at a time
by simpler instruments. Wavelength is oIten represented by the symbol . Similarly, Ior a
given substance, a standard graph oI the extinction coeIIicient (c) vs. wavelength () may be
made or used iI one is already available. Such a standard graph would be eIIectively
"concentration-corrected" and thus independent oI concentration. For the given substance, the
wavelength at which maximum absorption in the spectrum occurs is called
max
, pronounced
"Lambda-max".
The Woodward-Fieser rules rules are a set oI empirical observations which can be used to
predict
max
, the wavelength oI the most intense UV/Vis absorption, Ior conjugated organic
compounds such as dienes and ketones.
This spectrum can be used qualitatively to identiIy components in a sample as each
component has their own unique absorbance spectrum (like a Iingerprint).
INFRARED SPECTROSCOPY
Infrared spectroscopy (IR spectroscopy) is the subset oI spectroscopy that deals with the
inIrared region oI the electromagnetic spectrum. It covers a range oI techniques, the most
common being a Iorm oI absorption spectroscopy. As with all spectroscopic techniques, it
can be used to identiIy compounds or investigate sample composition. InIrared spectroscopy
correlation tables are tabulated in the literature.
1heory
The inIrared portion oI the electromagnetic spectrum is divided into three regions; the near-,
mid- and Iar- inIrared, named Ior their relation to the visible spectrum. The Iar-inIrared,
approximately 400-10 cm
-1
(100030 m), lying adjacent to the microwave region, has low
energy and may be used Ior rotational spectroscopy. The mid-inIrared, approximately 4000-
400 cm
-1
(301.4 m) may be used to study the Iundamental vibrations and associated
rotational-vibrational structure. The higher energy near-IR, approximately 14000-4000 cm
-1
(1.40.8 m) can excite overtone or harmonic vibrations. The names and classiIications oI
these subregions are merely conventions. They are neither strict divisions nor based on exact
molecular or electromagnetic properties.
InIrared spectroscopy exploits the Iact that molecules have speciIic Irequencies at which they
rotate or vibrate corresponding to discrete energy levels. These resonant Irequencies are
determined by the shape oI the molecular potential energy surIaces, the masses oI the atoms
and, by the associated vibronic coupling. In order Ior a vibrational mode in a molecule to be
IR active, it must be associated with changes in the permanent dipole. In particular, in the
Born-Oppenheimer and harmonic approximations, i.e. when the molecular Hamiltonian
corresponding to the electronic ground state can be approximated by a harmonic oscillator in
the neighborhood oI the equilibrium molecular geometry, the resonant Irequencies are
determined by the normal modes corresponding to the molecular electronic ground state
potential energy surIace. Nevertheless, the resonant Irequencies can be in a Iirst approach
related to the strength oI the bond, and the mass oI the atoms at either end oI it. Thus, the
Irequency oI the vibrations can be associated with a particular bond type.
Simple diatomic molecules have only one bond, which may stretch. More complex molecules
have many bonds, and vibrations can be conjugated, leading to inIrared absorptions at
characteristic Irequencies that may be related to chemical groups. For example, the atoms in a
CH
2
group, commonly Iound in organic compounds can vibrate in six diIIerent ways:
symmetrical and antisymmetrical stretching, scissoring, rocking, wagging and twisting:
The inIrared spectra oI a sample are collected by passing a beam oI inIrared light through the
sample. Examination oI the transmitted light reveals how much energy was absorbed at each
wavelength. This can be done with a monochromatic beam, which changes in wavelength
over time, or by using a Fourier transIorm instrument to measure all wavelengths at once.
From this, a transmittance or absorbance spectrum can be produced, showing at which IR
wavelengths the sample absorbs. Analysis oI these absorption characteristics reveals details
about the molecular structure oI the sample.
This technique works almost exclusively on samples with covalent bonds. Simple spectra are
obtained Irom samples with Iew IR active bonds and high levels oI purity. More complex
molecular structures lead to more absorption bands and more complex spectra. The technique
has been used Ior the characterization oI very complex mixtures.
Sample preparation
Gaseous samples require little preparation beyond puriIication, but a sample cell with a long
pathlength (typically 5-10 cm) is normally needed, as gases show relatively weak
absorbances.
Liquid samples can be sandwiched between two plates oI a high purity salt (commonly
sodium chloride, or common salt, although a number oI other salts such as potassium
bromide or calcium Iluoride are also used). The plates are transparent to the inIrared light and
will not introduce any lines onto the spectra. Some salt plates are highly soluble in water, so
the sample and washing reagents must be anhydrous (without water).
Solid samples can be prepared in two major ways. The Iirst is to crush the sample with a
mulling agent (usually nujol) in a marble or agate mortar, with a pestle. A thin Iilm oI the
mull is applied onto salt plates and measured.
The second method is to grind a quantity oI the sample with a specially puriIied salt (usually
potassium bromide) Iinely (to remove scattering eIIects Irom large crystals). This powder
mixture is then crushed in a mechanical die press to Iorm a translucent pellet through which
the beam oI the spectrometer can pass.
It is important to note that spectra obtained Irom diIIerent sample preparation methods will
look slightly diIIerent Irom each other due to diIIerences in the samples' physical states.
The last technique is the Cast Film technique.
Cast Iilm technique is used mainly Ior polymeric compound. Sample is Iirst dissolved in
suitable, non hygroscopic solvent. A drop oI this solution is deposited on surIace oI KBr or
NaCl cell. The solution is then evaporated to dryness and the Iilm Iormed on the cell is
analysed directly. Care is important to ensure that the Iilm is not too thick otherwise light
cannot pass through. This technique is suitable Ior qualitative analysis.
1ypical method
Typical apparatus
A beam oI inIrared light is produced and split into two separate beams. One is passed through
the sample, the other passed through a reIerence which is oIten the substance the sample is
dissolved in. The beams are both reIlected back towards a detector, however Iirst they pass
through a splitter which quickly alternates which oI the two beams enters the detector. The
two signals are then compared and a printout is obtained.
A reIerence is used Ior two reasons:
This prevents Iluctuations in the output oI the source aIIecting the data
This allows the eIIects oI the solvent to be cancelled out (the reIerence is usually a
pure Iorm oI the solvent the sample is in)
Uses and applications
InIrared spectroscopy is widely used in both research and industry as a simple and reliable
technique Ior measurement, quality control and dynamic measurement. The instruments are
now small, and can be transported, even Ior use in Iield trials. With increasing technology in
computer Iiltering and manipulation oI the results, samples in solution can now be measured
accurately (water produces a broad absorbance across the range oI interest, and thus renders
the spectra unreadable without this computer treatment). Some machines will also
automatically tell you what substance is being measured Irom a store oI thousands oI
reIerence spectra held in storage.
By measuring at a speciIic Irequency over time, changes in the character or quantity oI a
particular bond can be measured. This is especially useIul in measuring the degree oI
polymerization in polymer manuIacture. Modern research machines can take inIrared
measurements across the whole range oI interest as Irequently as 32 times a second. This can
be done whilst simultaneous measurements are made using other techniques. This makes the
observations oI chemical reactions and processes quicker and more accurate.
Techniques have been developed to assess the quality oI tea-leaves using inIrared
spectroscopy. This will mean that highly trained experts (also called 'noses') can be used
more sparingly, at a signiIicant cost saving.
|1|
InIrared spectroscopy has been highly successIul Ior applications in both organic and
inorganic chemistry. InIrared spectroscopy has also been successIully utilized in the Iield oI
semiconductor microelectronics
|2|
: Ior example, inIrared spectroscopy can be applied to
semiconductors like silicon, gallium arsenide, gallium nitride, zinc selenide, amorphous
silicon, silicon nitride, etc.
Isotope effects
The diIIerent isotopes in a particular species may give Iine detail in inIrared spectroscopy.
For example, the O-O stretching Irequency oI oxyhemocyanin is experimentally determined
to be 832 and 788 cm
-1
Ior v(
16
O-
16
O) and v(
18
O-
18
O) respectively.
By considering the O-O as a spring, the wavelength oI absorbance, v can be calculated:Where
k is the spring constant Ior the bond, and u is the reduced mass oI the A-B system:
(m
i
is the mass oI atom i).
The reduced masses Ior
16
O-
16
O and
18
O-
18
O can be approximated as 8 and 9 respectively.
Thus
Fourier transform infrared spectroscopy
Fourier transform infrared (FTIR) spectroscopy is a measurement technique Ior
collecting inIrared spectra. Instead oI recording the amount oI energy absorbed when the
Irequency oI the inIra-red light is varied (monochromator), the IR light is guided through an
interIerometer. AIter passing the sample the measured signal is the interIerogram. PerIorming
a mathematical Fourier transIorm on this signal results in a spectrum identical to that Irom
conventional (dispersive) inIrared spectroscopy.
FTIR spectrometers are cheaper than conventional spectrometers because building oI
interIerometers is easier than the Iabrication oI a monochromator. In addition, measurement
oI a single spectrum is Iaster Ior the FTIR technique because the inIormation at all
Irequencies is collected simultaneously. This allows multiple samples to be collected and
averaged together resulting in an improvement in sensitivity. Because oI its various
advantages, virtually all modern inIrared spectrometers are FTIR instruments.
1wo-dimensional infrared spectroscopy
Two-dimensional infrared correlation spectroscopy analysis is the application oI 2D
correlation analysis on inIrared spectra. By extending the spectral inIormation oI a perturbed
sample, spectral analysis is simpliIied and resolution is enhanced. The 2D synchronous and
2D asynchronous spectra represent a graphical overview oI the spectral changes due to a
perturbation (such as a changing concentration or changing temperature) as well as the
relationship between the spectral changes at two diIIerent wavenumbers.
Nonlinear two-dimensional infrared spectroscopy
|3||4|
is the inIrared version oI correlation
spectroscopy. Nonlinear two-dimensional inIrared spectroscopy is a technique that has
become available with the development oI Iemtosecond inIrared laser pulses. In this
experiment Iirst a set oI pump pulses are applied to the sample. This is Iollowed by a waiting
time, where the system is allowed to relax. The waiting time typically lasts Irom zero to
several picoseconds and the duration can be controlled with a resolution oI tens oI
Iemtoseconds. A probe pulse is then applied resulting in the emission oI a signal Irom the
sample. The nonlinear two-dimensional inIrared spectrum is a two-dimensional correlation
plot oI the Irequency e
1
that was excited by the initial pump pulses and the Irequency e
3
excited by the probe pulse aIter the waiting time. This allows the observation oI coupling
between diIIerent vibrational modes. Because oI its extremely high time resolution it can be
used to monitor molecular dynamics on a picosecond timescale. It is still a largely unexplored
technique and is becoming increasingly popular Ior Iundamental research.
Like in two-dimensional nuclear magnetic resonance (2DNMR) spectroscopy this technique
spreads the spectrum in two dimensions and allow Ior the observation oI cross peaks that
contain inIormation on the coupling between diIIerent modes. In contrast to 2DNMR
nonlinear two-dimensional inIrared spectroscopy also involve the excitation to overtones.
These excitations result in excited state absorption peaks located below the diagonal and
cross peaks. In 2DNMR two distinct techniques, COSY and NOESY, are Irequently used.
The cross peaks in the Iirst are related to the scalar coupling, while in the later they are
related to the spin transIer between diIIerent nuclei. In nonlinear two-dimensional inIrared
spectroscopy analogs have been drawn to these 2DNMR techniques. Nonlinear two-
dimensional inIrared spectroscopy with zero waiting time corresponds to COSY and
nonlinear two-dimensional inIrared spectroscopy with Iinite waiting time allowing
vibrational population transIer corresponds to NOESY. The COSY variant oI nonlinear two-
dimensional inIrared spectroscopy has been used Ior determination oI the secondary structure
content proteins.
|5|
RAMAN SPECTROSCOPY
Raman spectroscopy is a spectroscopic technique used in condensed matter physics and
chemistry to study vibrational, rotational, and other low-Irequency modes in a system.
|1|
It
relies on inelastic scattering, or Raman scattering oI monochromatic light, usually Irom a
laser in the visible, near inIrared, or near ultraviolet range. The laser light interacts with
phonons or other excitations in the system, resulting in the energy oI the laser photons being
shiIted up or down. The shiIt in energy gives inIormation about the phonon modes in the
system. InIrared spectroscopy yields similar, but complementary inIormation.
Typically, a sample is illuminated with a laser beam. Light Irom the illuminated spot is
collected with a lens and sent through a monochromator. Wavelengths close to the laser line,
due to elastic Rayleigh scattering, are Iiltered out while the rest oI the collected light is
dispersed onto a detector.
Spontaneous Raman scattering is typically very weak, and as a result the main diIIiculty oI
Raman spectroscopy is separating the weak inelastically scattered light Irom the intense
Rayleigh scattered laser light. Raman spectrometers typically use holographic diIIraction
gratings and multiple dispersion stages to achieve a high degree oI laser rejection. In the past,
PMTs were the detectors oI choice Ior dispersive Raman setups, which resulted in long
acquisition times. However, the recent uses oI CCD detectors have made dispersive Raman
spectral acquisition much more rapid.
Raman spectroscopy has a stimulated version, analogous to stimulated emission, called
stimulated Raman scattering.
Basic theory
Energy level diagram showing the states involved in Raman signal. The line thickness is
roughly proportional to the signal strength Irom the diIIerent transitions.
The Raman eIIect occurs when light impinges upon a molecule and interacts with the electron
cloud oI the bonds oI that molecule. The incident photon excites one oI the electrons into a
virtual state. For the spontaneous Raman eIIect, the molecule will be excited Irom the ground
state to a virtual energy state, and relax into a vibrational excited state, which generates
Stokes Raman scattering. II the molecule was already in an elevated vibrational energy state,
the Raman scattering is then called anti-Stokes Raman scattering.
A molecular polarizability change, or amount oI deIormation oI the electron cloud, with
respect to the vibrational coordinate is required Ior the molecule to exhibit the Raman eIIect.
The amount oI the polarizability change will determine the intensity, whereas the Raman shiIt
is equal to the vibrational level that is involved.
History
Although the inelastic scattering oI light was predicted by Smekal in 1923, it was not until
1928 that it was observed in practice. The Raman eIIect was named aIter one oI its
discoverers, the Indian scientist Sir C. V. Raman who observed the eIIect by means oI
sunlight (1928, together with K. S. Krishnan and independently by Grigory Landsberg and
Leonid Mandelstam).
|1|
Raman won the Nobel Prize in Physics in 1930 Ior this discovery
accomplished using sunlight, a narrow band photographic Iilter to create monochromatic light
and a "crossed" Iilter to block this monochromatic light. He Iound that light oI changed
Irequency passed through the "crossed" Iilter.
Subsequently the mercury arc became the principal light source, Iirst with photographic
detection and then with spectrophotometric detection. Currently lasers are used as light
sources.
Applications
Raman spectroscopy is commonly used in chemistry, since vibrational inIormation is very
speciIic Ior the chemical bonds in molecules. It thereIore provides a Iingerprint by which the
molecule can be identiIied. The Iingerprint region oI organic molecules is in the range 500-
2000 cm
-1
. Another way that the technique is used is to study changes in chemical bonding,
e.g. when a substrate is added to an enzyme.
Raman gas analyzers have many practical applications, Ior instance they are used in medicine
Ior real-time monitoring oI anaesthetic and respiratory gas mixtures during surgery.
In solid state physics, spontaneous Raman spectroscopy is used to, among other things,
characterize materials, measure temperature, and Iind the crystallographic orientation oI a
sample.
As with single molecules, a given solid material has characteristic phonon modes that can
help an experimenter identiIy it. In addition, Raman spectroscopy can be used to observe
other low Irequency excitations oI the solid, such as plasmons, magnons, and
superconducting gap excitations.
The spontaneous Raman signal gives inIormation on the population oI a given phonon mode
in the ratio between the Stokes (downshiIted) intensity and anti-Stokes (upshiIted) intensity.
Raman scattering by an anisotropic crystal gives inIormation on the crystal orientation. The
polarization oI the Raman scattered light with respect to the crystal and the polarization oI the
laser light can be used to Iind the orientation oI the crystal, iI the crystal structure
(speciIically, its point group) is known.
Raman active Iibers, such as aramid and carbon, have vibrational modes that show a shiIt in
Raman Irequency with applied stress. Polypropylene Iibers also exhibit similar shiIts.
The radial breathing mode is a commonly used technique to evaluate the diameter oI carbon
nanotubes.
Spatially OIIset Raman Spectroscopy (SORS), which is less sensitive to surIace layers than
conventional Raman, can be used to discover counterIeit drugs without opening their internal
packaging, and Ior non-invasive monitoring oI biological tissue.
|2||3|
Raman spectroscopy can be used to investigate the chemical composition oI historical
documents such as the Book oI Kells and contribute to knowledge oI the social and economic
conditions at the time the documents were produced.
|4|
This is especially helpIul because
Raman spectroscopy oIIers a non-invasive way to determine the best course oI preservation
or conservation treatment Ior such materials.
Raman microspectroscopy
Raman spectroscopy oIIers several advantages Ior microscopic analysis. Since it is a
scattering technique, specimens do not need to be Iixed or sectioned. Raman spectra can be
collected Irom a very small volume ( 1 m in diameter); these spectra allow the
identiIication oI species present in that volume. Water does not interIere very strongly. Thus,
Raman spectroscopy is suitable Ior the microscopic examination oI minerals, materials such
as polymers and ceramics, cells and proteins. A Raman microscope begins with a standard
optical microscope, and adds an excitation laser, a monochromator, and a sensitive detector
(such as a charge-coupled device (CCD) or photomultiplier tube (PMT)). FT-Raman has also
been used with microscopes.
In direct imaging, the whole Iield oI view is examined Ior scattering over a small range oI
wavenumbers (Raman shiIts). For instance, a wavenumber characteristic Ior cholesterol could
be used to record the distribution oI cholesterol within a cell culture.
The other approach is hvperspectral imaging or chemical imaging, in which thousands oI
Raman spectra are acquired Irom all over the Iield oI view. The data can then be used to
generate images showing the location and amount oI diIIerent components. Taking the cell
culture example, a hyperspectral image could show the distribution oI cholesterol, as well as
proteins, nucleic acids, and Iatty acids. Sophisticated signal- and image-processing
techniques can be used to ignore the presence oI water, culture media, buIIers, and other
interIerents.
Raman microscopy, and in particular conIocal microscopy, has very high spatial resolution.
For example, the lateral and depth resolutions were 250 nm and 1.7 m, respectively, using a
conIocal Raman microspectrometer with the 632.8 nm line Irom a He-Ne laser with a pinhole
oI 100 m diameter.
Since the objective lenses oI microscopes Iocus the laser beam to several micrometres in
diameter, the resulting photon Ilux is much higher than achieved in conventional Raman
setups. This has the added beneIit oI enhanced Iluorescence quenching. However, the high
photon Ilux can also cause sample degradation, and Ior this reason some setups require a
thermally conducting substrate (which acts as a heat sink) in order to mitigate this process.
By using Raman microspectroscopy, in vivo time- and space-resolved Raman spectra oI
microscopic regions oI samples can be measured. As a result, the Iluorescence oI water,
media, and buIIers can be removed. Consequently in vivo time- and space-resolved Raman
spectroscopy is suitable to examine proteins, cells and organs.
Raman microscopy Ior biological and medical specimens generally uses near-inIrared (NIR)
lasers (785 nm diodes and 1064 nm Nd:YAG are especially common). This reduces the risk
oI damaging the specimen by applying high power. However, the intensity oI NIR Raman is
low (owing to the e
-4
dependence oI Raman scattering intensity), and most detectors required
very long collection times. Recently, more sensitive detectors have become available, making
the technique better suited to general use. Raman microscopy oI inorganic specimens, such as
rocks and ceramics and polymers, can use a broader range oI excitation wavelengths.
|5|
Jariations
Several variations oI Raman spectroscopy have been developed. The usual purpose is to
enhance the sensitivity (e.g., surIace-enhanced Raman), to improve the spatial resolution
(Raman microscopy), or to acquire very speciIic inIormation (resonance Raman).
Surface Enhanced Raman Spectroscopy (SERS) - Normally done in a silver or
gold colloid or a substrate containing silver or gold. SurIace plasmons oI silver and
gold are easily excited by the laser, and the resulting electric Iields cause other nearby
molecules to become Raman active. The result is ampliIication oI the Raman signal
(by up to 10
11
). This eIIect was originally observed by Fleishman but the prevailing
explanation was proposed by Van Duyne in 1977.
|6|
Hyper Raman - A non-linear eIIect in which the vibrational modes interact with the
second harmonic oI the excitation beam. This requires very high power, but allows
the observation oI vibrational modes which are normally "silent". It Irequently relies
on SERS-type enhancement to boost the sensitivity.
Resonance Raman spectroscopy - The excitation wavelength is matched to an
electronic transition oI the molecule or crystal, so that vibrational modes associated
with the excited electronic state are greatly enhanced. This is useIul Ior studying large
molecules such as polypeptides, which might show hundreds oI bands in
"conventional" Raman spectra. It is also useIul Ior associating normal modes with
their observed Irequency shiIts.
Spontaneous Raman Spectroscopy - Used to study the temperature dependence oI
the Raman spectra oI molecules.
Optical Tweezers Raman Spectroscopy (OTRS) - Used to study individual
particles, and even biochemical processes in single cells trapped by optical tweezers.
Stimulated Raman Spectroscopy - A two color pulse transIers the population Irom
ground to a rovibrationally excited state, iI the diIIerence in energy corresponds to an
allowed Raman transition. Two photon UV ionization, applied aIter the population
transIer but beIore relaxation, allows the intra-molecular or inter-molecular Raman
spectrum oI a gas or molecular cluster (indeed, a given conIormation oI molecular
cluster) to be collected. This is a useIul molecular dynamics technique.
Spatially Offset Raman Spectroscopy (SORS) - The Raman scatter is collected
Irom regions laterally oIIset away Irom the excitation laser spot, leading to
signiIicantly lower contributions Irom the surIace layer than with traditional Raman
spectroscopy.
UNIT IV
THERMAL ANALYSIS
THERMOGRAVIMETRIC ANALYSIS
sketch oI a typical TGA (Setaram TG-DTA 92 B type); the cooling water pipe was omitted
Thermogravimetric Analysis or TGA is a type oI testing that is perIormed on samples to
determine changes in weight in relation to change in temperature. Such analysis relies on a
high degree oI precision in three measurements: weight, temperature, and temperature
change. As many weight loss curves look similar, the weight loss curve may require
transIormation beIore results may be interpreted. A derivative weight loss curve can be used
to tell the point at which weight loss is most apparent. Again, interpretation is limited without
Iurther modiIications and deconvolution oI the overlapping peaks may be required.
TGA is commonly employed in research and testing to determine characteristics oI materials
such as polymers, to determine degradation temperatures, absorbed moisture content oI
materials, the level oI inorganic and organic components in materials, decomposition points
oI explosives, and solvent residues. It is also oIten used to estimate the corrosion kinetics in
high temperature oxidation.
Analyzer
The analyzer usually consists oI a high-precision balance with a pan loaded with the sample.
The sample is placed in a small electrically heated oven with a thermocouple to accurately
measure the temperature. The atmosphere may be purged with an inert gas to prevent
oxidation or other undesired reactions. A computer is used to control the instrument.
Analysis is carried out by raising the temperature gradually and plotting weight against
temperature. AIter the data is obtained, curve smoothing and other operations may be done
such as to Iind the exact points oI inIlection.
DIFFERENTIAL THERMAL ANALYSIS
Differential thermal analysis (or DTA) is a thermoanalytic technique, similar to diIIerential
scanning calorimetry. In DTA, the material under study and an inert reIerence are heated (or
cooled) under identical conditions, while recording any temperature diIIerence between
sample and reIerence.
|1|
This diIIerential temperature is then plotted against time, or against
temperature (DTA curve or thermogram). Changes in the sample, either exothermic or
endothermic, can be detected relative to the inert reIerence. Thus, a DTA curve provides data
on the transIormations that have occurred, such as glass transitions, crystallization, melting
and sublimation. The area under a DTA peak can be to the enthalpy change and it is not
aIIected by the heat capacity oI the sample.
Apparatus
A DTA apparatus consist oI a sample holder comprising thermocouples, sample containers
and a ceramic or metallic block; a Iurnace; a temperature programmer; and a recording
system. The key Ieature is the existence oI two thermocouples connected to a voltmeter. One
thermocouple is placed in an inert material such as Al2O3, while the other is placed in a
sample oI the material under study. As the temperature is increased, there will be a brieI
deIlection oI the voltmeter iI the sample is undergoing a phase transition. This occurs because
the input oI heat will raise the temperature oI the inert substance, but be incorporated as latent
heat in the material changing phase.
Applications
A DTA curve can be used only as a finger print Ior identiIication purposes but usually the
applications oI this method are the determination oI phase diagrams, heat change
measurements and decomposition in various atmospheres.
DTA is widely used in the pharmaceutical and Iood industries.
DTA may be used in cement chemistry, mineralogical research and in environmental studies.
DTA curves may also be used to date bone remains or to study archaeological materials.
DIFFERENTIAL SCANNING CALORIMETRY
Differential scanning calorimetry or DSC is a thermoanalytical technique in which the
diIIerence in the amount oI heat required to increase the temperature oI a sample and
reIerence are measured as a Iunction oI temperature. Both the sample and reIerence are
maintained at nearly the same temperature throughout the experiment. Generally, the
temperature program Ior a DSC analysis is designed such that the sample holder temperature
increases linearly as a Iunction oI time. The reIerence sample should have a well-deIined heat
capacity over the range oI temperatures to be scanned. The basic principle underlying this
technique is that, when the sample undergoes a physical transIormation such as phase
transitions, more (or less) heat will need to Ilow to it than the reIerence to maintain both at
the same temperature. Whether more or less heat must Ilow to the sample depends on
whether the process is exothermic or endothermic. For example, as a solid sample melts to a
liquid it will require more heat Ilowing to the sample to increase its temperature at the same
rate as the reIerence. This is due to the absorption oI heat by the sample as it undergoes the
endothermic phase transition Irom solid to liquid. Likewise, as the sample undergoes
exothermic processes (such as crystallization) less heat is required to raise the sample
temperature. By observing the diIIerence in heat Ilow between the sample and reIerence,
diIIerential scanning calorimeters are able to measure the amount oI heat absorbed or
released during such transitions. DSC may also be used to observe more subtle phase
changes, such as glass transitions. DSC is widely used in industrial settings as a quality
control instrument due to its applicability in evaluating sample purity and Ior studying
polymer curing.
|1||2||3|
An alternative technique, which shares much in common with DSC, is diIIerential thermal
analysis (DTA). In this technique it is the heat Ilow to the sample and reIerence that remains
the same rather than the temperature. When the sample and reIerence are heated identically
phase changes and other thermal processes cause a diIIerence in temperature between the
sample and reIerence. Both DSC and DTA provide similar inIormation; DSC is the more
widely used oI the two techniques.
|1||2||3|
DSC curves
The result oI a DSC experiment is a curve oI heat Ilux versus temperature or versus time.
There are two diIIerent conventions: exothermic reactions in the sample shown with a
positive or negative peak; it depends by the diIIerent kind oI technology used by the
instrumentation to make the experiment. This curve can be used to calculate enthalpies oI
transitions. This is done by integrating the peak corresponding to a given transition. It can be
shown that the enthalpy oI transition can be expressed using the Iollowing equation:
AH KA
where AH is the enthalpy oI transition, K is the calorimetric constant, and A is the area under
the curve. The calometric constant will vary Irom instrument to instrument, and can be
determined by analyzing a well-characterized sample with known enthalpies oI transition.
|2|
Applications
DiIIerential scanning calorimetry can be used to measure a number oI characteristic
properties oI a sample. Using this technique it is possible to observe Iusion and crystallization
events as well as glass transition temperatures (T
g
). DSC can also be used to study oxidation,
as well as other chemical reactions.
|1||2||3|
Glass transitions may occur as the temperature oI an amorphous solid is increased. These
transitions appear as a step in the baseline oI the recorded DSC signal. This is due to the
sample undergoing a change in heat capacity; no Iormal phase change occurs.
|1||3|
As the temperature increases, an amorphous solid will become less viscous. At some point
the molecules may obtain enough Ireedom oI motion to spontaneously arrange themselves
into a crystalline Iorm. This is known as the crystallization temperature (T
c
). This transition
Irom amorphous solid to crystalline solid is an exothermic process, and results in a peak in
the DSC signal. As the temperature increases the sample eventually reaches its melting
temperature (T
m
). The melting process results in an endothermic peak in the DSC curve. The
ability to determine transition temperatures and enthalpies makes DSC an invaluable tool in
producing phase diagrams Ior various chemical systems.
DSC may also be used in the study oI liquid crystals. As matter transitions between solid and
liquid it oIten goes through a third state, which displays properties oI both phases. This
anisotropic liquid is known as a liquid crystalline or mesomorphous state. Using DSC, it is
possible to observe the small energy changes that occur as matter transitions Irom a solid to a
liquid crystal and Irom a liquid crystal to an isotropic liquid.
|2|
Using diIIerential scanning calorimetry to study the oxidative stability oI samples generally
requires an airtight sample chamber. Usually, such tests are done isothermally (at constant
temperature) by changing the atmosphere oI the sample. First, the sample is brought to the
desired test temperature under an inert atmosphere, usually nitrogen. Then, oxygen is added
to the system. Any oxidation that occurs is observed as a deviation in the baseline. Such
analyses can be used to determine the stability and optimum storage conditions Ior a
compound.
|1|
DSC is widely used in the pharmaceutical and polymer industries. For the polymer chemist,
DSC is a handy tool Ior studying curing processes, which allows the Iine tuning oI polymer
properties. The cross-linking oI polymer molecules that occurs in the curing process is
exothermic, resulting in a positive peak in the DSC curve that usually appears soon aIter the
glass transition.
|1||2||3|
In the pharmaceutical industry it is necessary to have well-characterized drug compounds in
order to deIine processing parameters. For instance, iI it is necessary to deliver a drug in the
amorphous Iorm, it is desirable to process the drug at temperatures below those at which
crystallization can occur.
In Iood science research, DSC is used in conjunction with other thermal analytical techniques
to determine water dynamics. Changes in water distribution may be correlated with changes
in texture. Similar to material science studies, the eIIects oI curing on conIectionery products
can also be analyzed.
DSC curves may also be used to evaluate drug and polymer purities. This is possible because
the temperature range over which a mixture oI compounds melts is dependent on their
relative amounts. This eIIect is due to a phenomenon known as Ireezing point depression,
which occurs when a Ioreign solute is added to a solution. (Freezing point depression is what
allows salt to de-ice sidewalks and antiIreeze to keep your car running in the winter.)
Consequently, less pure compounds will exhibit a broadened melting peak that begins at
lower temperature than a pure compound.
In last Iew years this technology has been involved in metallic material study. The
characterization oI this kind oI material with DSC is not easy yet because oI the low quantity
oI literature about it. It is known that it is possible to use DSC to Iind solidus and liquidus
temperature oI a metal alloy, but the widest application is, by now, the study oI
precipitations, Guiner Preston zones, phase transitions, dislocations movement, grain growth
etc.
UNIT V
SEPARATION TECHNIQUES
INTRODUCTION
Chromatography (Irom Greek peu:chroma, color and puciv:"graphein" to write) is the
collective term Ior a Iamily oI laboratory techniques Ior the separation oI mixtures. It
involves passing a mixture dissolved in a "mobile phase" through a stationarv phase, which
separates the analyte to be measured Irom other molecules in the mixture and allows it to be
isolated.
Chromatography may be preparative or analytical. Preparative chromatography seeks to
separate the components oI a mixture Ior Iurther use (and is thus a Iorm oI puriIication).
Analytical chromatography normally operates with smaller amounts oI material and seeks to
measure the relative proportions oI analytes in a mixture. The two are not mutually exclusive.
Explanation
.An analogy which is sometimes useIul is to suppose a mixture oI bees and wasps passing
over a Ilower bed. The bees would be more attracted to the Ilowers than the wasps, and
would become separated Irom them. II one were to observe at a point past the Ilower bed, the
wasps would pass Iirst, Iollowed by the bees. In this analogy, the bees and wasps represent
the analytes to be separated, the Ilowers represent the stationary phase, and the mobile phase
could be thought oI as the air. The key to the separation is the diIIering aIIinities among
analyte, stationary phase, and mobile phase. The observer could represent the detector used in
some Iorms oI analytical chromatography. A key point is that the detector need not be
capable oI discriminating between the analytes, since they have become separated beIore
passing the detector.
Chromatography terms
The analyte is the substance that is to be separated during chromatography.
Analytical chromatography is used to determine the existence and possibly also the
concentration oI analyte(s) in a sample.
A bonded phase is a stationary phase that is covalently bonded to the support
particles or to the inside wall oI the column tubing.
A chromatogram is the visual output oI the chromatograph. In the case oI an optimal
separation, diIIerent peaks or patterns on the chromatogram correspond to diIIerent
components oI the separated mixture.
Plotted on the x-axis is the retention time and plotted on the y-axis a signal (Ior
example obtained by a spectrophotometer, mass spectrometer or a variety oI other
detectors) corresponding to the response created by the analytes exiting the system. In
the case oI an optimal system the signal is proportional to the concentration oI the
speciIic analyte separated.
A chromatograph is equipment that enables a sophisticated separation e.g. gas
chromatographic or liquid chromatographic separation.
Chromatography is a physical method oI separation in which the components to be
separated are distributed between two phases, one oI which is stationary (stationary
phase) while the other (the mobile phase) moves in a deIinite direction.
The effluent is the mobile phase leaving the column.
An immobilized phase is a stationary phase which is immobilized on the support
particles, or on the inner wall oI the column tubing.
The mobile phase is the phase which moves in a deIinite direction. It may be a liquid
(LC and CEC), a gas (GC), or a supercritical Iluid (supercritical-Iluid
chromatography, SFC). A better deIinition: The mobile phase consists oI the sample
being separated/analyzed and the solvent that moves the sample through the column.
In one case oI HPLC the solvent consists oI a carbonate/bicarbonate solution and the
sample is the anions being separated. The mobile phase moves through the
chromatography column (the stationary phase) where the sample interacts with the
stationary phase and is separated.
Preparative chromatography is used to puriIy suIIicient quantities oI a substance
Ior Iurther use, rather than analysis.
The retention time is the characteristic time it takes Ior a particular analyte to pass
through the system (Irom the column inlet to the detector) under set conditions. See
also: Kovat's retention index
The sample is the matter analysed in chromatography. It may consist oI a single
component or it may be a mixture oI components. When the sample is treated in the
course oI an analysis, the phase or the phases containing the analytes oI interest is/are
reIerred to as the sample whereas everything out oI interest separated Irom the sample
beIore or in the course oI the analysis is reIerred to as waste.
The solute reIers to the sample components in partition chromatography.
The solvent reIers to any substance capable oI solubilizing other substance, and
especially the liquid mobile phase in LC.
The stationary phase is the substance which is Iixed in place Ior the chromatography
procedure. Examples include the silica layer in thin layer chromatography.
Techniques by chromatographic bed shape
Column chromatography
A diagram oI a standard column chromatography and a Ilash column chromatography setup
Column chromatography is a separation technique in which the stationary bed is within a
tube. The particles oI the solid stationary phase or the support coated with a liquid stationary
phase may Iill the whole inside volume oI the tube (packed column) or be concentrated on or
along the inside tube wall leaving an open, unrestricted path Ior the mobile phase in the
middle part oI the tube (open tubular column). DiIIerences in rates oI movement through the
medium are calculated to diIIerent retention times oI the sample.
In 1978, W. C. Still introduced a modiIied version oI column chromatography called flash
column chromatography (Ilash). The technique is very similar to the traditional column
chromatography, except Ior that the solvent is driven through the column by applying
positive pressure. This allowed most separations to be perIormed in less than 20 minutes,
with improved separations compared to the old method. Modern Ilash chromatography
systems are sold as pre-packed plastic cartridges, and the solvent is pumped through the
cartridge. Systems may also be linked with detectors and Iraction collectors providing
automation. The introduction oI gradient pumps resulted in quicker separations and less
solvent usage.
In expanded bed adsorption, a Iluidized bed is used, rather than a solid phase made by a
packed bed. This allows omission oI initial clearing steps such as centriIugation and
Iiltration, Ior culture broths or slurries oI broken cells.
Planar Chromatography
Thin layer chromatography is used to separate components oI chlorophyll
Planar chromatography is a separation technique in which the stationary phase is present as
or on a plane. The plane can be a paper, serving as such or impregnated by a substance as the
stationary bed (paper chromatography) or a layer oI solid particles spread on a support such
as a glass plate (thin layer chromatography).
Paper Chromatography
Paper chromatography is a technique that involves placing a small dot oI sample solution
onto a strip oI chromatography paper. The paper is placed in a jar containing a shallow layer
oI solvent and sealed. As the solvent rises through the paper it meets the sample mixture
which starts to travel up the paper with the solvent. DiIIerent compounds in the sample
mixture travel diIIerent distances according to how strongly they interact with the paper. This
paper is made oI cellulose, a polar molecule, and the compounds within the mixture travel
Iarther iI they are non-polar. More polar substances bond with the cellulose paper more
quickly, and thereIore do not travel as Iar. This process allows the calculation oI an R
I
value
and can be compared to standard compounds to aid in the identiIication oI an unknown
substance.
Thin layer chromatography
Thin layer chromatography (TLC) is a widely-employed laboratory technique and is similar
to paper chromatography. However, instead oI using a stationary phase oI paper, it involves a
stationary phase oI a thin layer oI adsorbent like silica gel, alumina, or cellulose on a Ilat,
inert substrate. Compared to paper, it has the advantage oI Iaster runs, better separations, and
the choice between diIIerent adsorbents. DiIIerent compounds in the sample mixture travel
diIIerent distances according to how strongly they interact with the adsorbent. This allows the
calculation oI an R
I
value and can be compared to standard compounds to aid in the
identiIication oI an unknown substance.
Techniques by physical state of mobile phase
Gas chromatography
Gas chromatography (GC), also sometimes known as Gas-Liquid chromatography, (GLC), is
a separation technique in which the mobile phase is a gas. Gas chromatography is always
carried out in a column, which is typically "packed" or "capillary" (see below) .
Gas chromatography (GC) is based on a partition equilibrium oI analyte between a solid
stationary phase (oIten a liquid silicone-based material) and a mobile gas (most oIten
Helium). The stationary phase is adhered to the inside oI a small-diameter glass tube (a
capillary column) or a solid matrix inside a larger metal tube (a packed column). It is widely
used in analytical chemistry; though the high temperatures used in GC make it unsuitable Ior
high molecular weight biopolymers or proteins (heat will denature them), Irequently
encountered in biochemistry, it is well suited Ior use in the petrochemical, environmental
monitoring, and industrial chemical Iields. It is also used extensively in chemistry research.
Liquid chromatography
Liquid chromatography (LC) is a separation technique in which the mobile phase is a liquid.
Liquid chromatography can be carried out either in a column or a plane. Present day liquid
chromatography that generally utilizes very small packing particles and a relatively high
pressure is reIerred to as high perIormance liquid chromatography (HPLC).
In the HPLC technique, the sample is Iorced through a column that is packed with irregularly
or spherically shaped particles or a porous monolithic layer (stationary phase) by a liquid
(mobile phase) at high pressure. HPLC is historically divided into two diIIerent sub-classes
based on the polarity oI the mobile and stationary phases. Technique in which the stationary
phase is more polar than the mobile phase (e.g. toluene as the mobile phase, silica as the
stationary phase) is called normal phase liquid chromatography (NPLC) and the opposite
(e.g. water-methanol mixture as the mobile phase and C18 octadecylsilyl as the stationary
phase) is called reversed phase liquid chromatography (RPLC). Ironically the "normal phase"
has Iewer applications and RPLC is thereIore used considerably more.
SpeciIic techniques which come under this broad heading are listed below. It should also be
noted that the Iollowing techniques can also be considered Iast protein liquid chromatography
iI no pressure is used to drive the mobile phase through the stationary phase. See also
Aqueous Normal Phase Chromatography.
Affinity chromatography
AIIinity chromatography is based on selective non-covalent interaction between an analyte
and speciIic molecules. It is very speciIic, but not very robust. It is oIten used in biochemistry
in the puriIication oI proteins bound to tags. These Iusion proteins are labelled with
compounds such as His-tags, biotin or antigens, which bind to the stationary phase
speciIically. AIter puriIication, some oI these tags are usually removed and the pure protein is
obtained.
Supercritical fluid chromatography
Supercritical Iluid chromatography is a separation technique in which the mobile phase is a
Iluid above and relatively close to its critical temperature and pressure.
Techniques by separation mechanism
Ion exchange chromatography
Ion exchange chromatography utilizes ion exchange mechanism to separate analytes. It is
usually perIormed in columns but the mechanism can be beneIited also in planar mode. Ion
exchange chromatography uses a charged stationary phase to separate charged compounds
including amino acids, peptides, and proteins. In conventional methods the stationary phase is
an ion exchange resin that carries charged Iunctional groups which interact with oppositely
charged groups oI the compound to be retained. Ion exchange chromatography is commonly
used to puriIy proteins using FPLC.
Size exclusion chromatography
Size exclusion chromatography (SEC) is also known as gel permeation chromatography
(GPC) or gel filtration chromatography and separates molecules according to their size (or
more accurately according to their hydrodynamic diameter or hydrodynamic volume).
Smaller molecules are able to enter the pores oI the media and, thereIore, take longer to elute,
whereas larger molecules are excluded Irom the pores and elute Iaster. It is generally a low
resolution chromatography technique and thus it is oIten reserved Ior the Iinal, "polishing"
step oI puriIication. It is also useIul Ior determining the tertiary structure and quaternary
structure oI puriIied proteins, especially since it can be carried out under native solution
conditions.
Special techniques
Reversed-phase chromatography
Reversed-phase chromatography is an elution procedure used in liquid chromatography in
which the mobile phase is signiIicantly more polar than the stationary phase.
Two-dimensional chromatography
In some cases, the chemistry within a given column can be insuIIicient to separate some
analytes. It is possible to direct a series oI unresolved peaks onto a second column with
diIIerent physico-chemical (Chemical classiIication) properties. Since the mechanism oI
retention on this new solid support is diIIerent Irom the Iirst dimensional separation, it can be
possible to separate compounds that are indistinguishable by one-dimensional
chromatography.
Pyrolysis gas chromatography
Fast protein liquid chromatography
Fast protein liquid chromatography (FPLC) is a term applied to several chromatography
techniques which are used to puriIy proteins. Many oI these techniques are identical to those
carried out under high perIormance liquid chromatography.
Countercurrent chromatography
Countercurrent chromatography (CCC) is a type oI liquid-liquid chromatography, where both
the stationary and mobile phases are liquids. It involves mixing a solution oI liquids, allowing
them to settle into layers and then separating the layers.
Chiral chromatography
Chiral chromatography involves the separation oI stereoisomers. In the case oI enantiomers,
these have no chemical or physical diIIerences apart Irom being three dimensional mirror
images. Conventional chromatography or other separation processes are incapable oI
separating them. To enable chiral separations to take place, either the mobile phase or the
stationary phase must themselves be made chiral, giving diIIering aIIinities between the
analytes. Chiral chromatography HPLC columns (with a chiral stationary phase) in both
normal and reversed phase are commercially available
Potrebbero piacerti anche
- Instrument Control Uning LABVIEWDocumento69 pagineInstrument Control Uning LABVIEWcoolhemakumarNessuna valutazione finora
- Instrumental Analysis Lecture Notes IIIDocumento62 pagineInstrumental Analysis Lecture Notes IIIcoolhemakumar100% (1)
- Electromagnetic RadiationDocumento2 pagineElectromagnetic RadiationcoolhemakumarNessuna valutazione finora
- Interfacing To Microprocessor ControllerDocumento14 pagineInterfacing To Microprocessor ControllercoolhemakumarNessuna valutazione finora
- Instrumental Analysis Lecture Notes IIDocumento56 pagineInstrumental Analysis Lecture Notes IIcoolhemakumar100% (3)
- V / F ConverterDocumento8 pagineV / F ConvertercoolhemakumarNessuna valutazione finora
- Interaction of EM Radiation and MatterDocumento3 pagineInteraction of EM Radiation and MattercoolhemakumarNessuna valutazione finora
- Basics of V/F and F/V ConvertersDocumento11 pagineBasics of V/F and F/V ConverterscoolhemakumarNessuna valutazione finora
- LabVIEW IntroductionDocumento89 pagineLabVIEW Introductioncoolhemakumar100% (1)
- IR Spectroscopy BasicsDocumento16 pagineIR Spectroscopy BasicscoolhemakumarNessuna valutazione finora
- Virtual Instrumentation ArchitectureDocumento25 pagineVirtual Instrumentation Architecturecoolhemakumar100% (8)
- Unit 7 Flame PhotometryDocumento32 pagineUnit 7 Flame PhotometryRia Agnez100% (8)
- Mass SpectrometryDocumento19 pagineMass SpectrometrycoolhemakumarNessuna valutazione finora
- Displacement and PositionDocumento6 pagineDisplacement and PositioncoolhemakumarNessuna valutazione finora
- NMR Spectroscopy - Short NoteDocumento6 pagineNMR Spectroscopy - Short Notecoolhemakumar100% (4)
- Molecular Energy Levels and IR SpectrosDocumento27 pagineMolecular Energy Levels and IR SpectroscoolhemakumarNessuna valutazione finora
- Energy Levels and Transitions in AtomsDocumento7 pagineEnergy Levels and Transitions in AtomscoolhemakumarNessuna valutazione finora
- Automation in DVMsDocumento7 pagineAutomation in DVMscoolhemakumarNessuna valutazione finora
- Electron Spin ResonanceDocumento29 pagineElectron Spin Resonancecoolhemakumar100% (2)
- Digital and Pulse-Train Conditioning: Digital I/O InterfacingDocumento6 pagineDigital and Pulse-Train Conditioning: Digital I/O InterfacingcoolhemakumarNessuna valutazione finora
- NMR SpectrometerDocumento13 pagineNMR SpectrometercoolhemakumarNessuna valutazione finora
- NMRDocumento142 pagineNMRMaulana Senjaya SusiloNessuna valutazione finora
- Mass SpecDocumento38 pagineMass SpecRashi VermaNessuna valutazione finora
- Mass Spectrometry - Short NoteDocumento2 pagineMass Spectrometry - Short NotecoolhemakumarNessuna valutazione finora
- CASE ToolsDocumento5 pagineCASE ToolscoolhemakumarNessuna valutazione finora
- Hart TutorialDocumento6 pagineHart TutorialabhiklNessuna valutazione finora
- Virtual Instrumentation SystemsDocumento7 pagineVirtual Instrumentation SystemscoolhemakumarNessuna valutazione finora
- What Is Modular InstrumentationDocumento7 pagineWhat Is Modular InstrumentationcoolhemakumarNessuna valutazione finora
- AN713 - Controller Area Network (CAN) BasicsDocumento9 pagineAN713 - Controller Area Network (CAN) BasicsRam Krishan SharmaNessuna valutazione finora
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (399)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (73)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Physics Enrich Test STPM Sem 1Documento17 paginePhysics Enrich Test STPM Sem 1雷顺开Nessuna valutazione finora
- Assignment N1Documento9 pagineAssignment N1Rania ChNessuna valutazione finora
- Chemistry Quiz Chapter 4 Form 4 @Documento3 pagineChemistry Quiz Chapter 4 Form 4 @Mohd Norihwan100% (1)
- Raksasa CVDocumento5 pagineRaksasa CVLydia Febriana SimanjuntakNessuna valutazione finora
- FORNEY TestingMachinesDocumento17 pagineFORNEY TestingMachinesNhayelli EstradaNessuna valutazione finora
- Practical EXamDocumento5 paginePractical EXamNDTInstructor100% (6)
- Csi ReferDocumento502 pagineCsi Referrenzo wilber bernedo beltranNessuna valutazione finora
- Mathematics in The Modern WorldDocumento15 pagineMathematics in The Modern WorldMikaela MelegritoNessuna valutazione finora
- E 777 - Gidarakos 2005 - Ultimate - C and HDocumento5 pagineE 777 - Gidarakos 2005 - Ultimate - C and HTamara Aulia RachimNessuna valutazione finora
- AluminioDocumento14 pagineAluminiobrauliocoroNessuna valutazione finora
- Travelling Tripper CalculationDocumento5 pagineTravelling Tripper CalculationHarshGuptaNessuna valutazione finora
- Metering - Manual - Eastern - RegionDocumento65 pagineMetering - Manual - Eastern - RegionAmit PathakNessuna valutazione finora
- Aaronia AARTOS DDS FAQ PDFDocumento10 pagineAaronia AARTOS DDS FAQ PDFUmar FarooqNessuna valutazione finora
- Crystallinity in Polymers-1Documento19 pagineCrystallinity in Polymers-1anbuchelvanNessuna valutazione finora
- Sample Pure PaperDocumento28 pagineSample Pure PapersaNessuna valutazione finora
- Full Text of "Waterproofing EngineeringDocumento966 pagineFull Text of "Waterproofing Engineeringmazharul43Nessuna valutazione finora
- Gas BookDocumento202 pagineGas Bookomiitg67% (3)
- BENNET 840 MallinckrodtDocumento12 pagineBENNET 840 MallinckrodtmartynbbNessuna valutazione finora
- Grade Xii Book III FinalDocumento158 pagineGrade Xii Book III FinalEdwinNessuna valutazione finora
- The Key To Super Consciousness Chapter 1Documento6 pagineThe Key To Super Consciousness Chapter 1Will FortuneNessuna valutazione finora
- Lutensol XP Surfactants Nonionic Surfactants For DetergentsDocumento11 pagineLutensol XP Surfactants Nonionic Surfactants For DetergentsKaran Kumar VermaNessuna valutazione finora
- Banner Engineering - Glass Fiber Series - CatalogDocumento43 pagineBanner Engineering - Glass Fiber Series - CatalogTavo CoxNessuna valutazione finora
- Discrete-Time Fourier Transform: XN X XneDocumento36 pagineDiscrete-Time Fourier Transform: XN X Xneyadavsticky5108Nessuna valutazione finora
- Cfy CRP (2125) - pt-2 B Lot - Iitjee Stage-RDocumento16 pagineCfy CRP (2125) - pt-2 B Lot - Iitjee Stage-RRitvik RajNessuna valutazione finora
- Presentation Dr. Fahmida Gulshan 1Documento30 paginePresentation Dr. Fahmida Gulshan 1Vishal SharmaNessuna valutazione finora
- Bio Analytical SopDocumento17 pagineBio Analytical SopalexpharmNessuna valutazione finora
- Trig Crash CourseDocumento28 pagineTrig Crash CourseHimraj BachooNessuna valutazione finora
- Multi Meter Triplett 630-NA Tested by ZS1JHGDocumento2 pagineMulti Meter Triplett 630-NA Tested by ZS1JHGJohn Howard GreenNessuna valutazione finora
- SSPC Pa 2Documento16 pagineSSPC Pa 2Rony Ruiz100% (5)
- States of Matter Lesson PlanDocumento8 pagineStates of Matter Lesson Planapi-383721875Nessuna valutazione finora