Documenti di Didattica
Documenti di Professioni
Documenti di Cultura
Voltametria e Polarografia
Caricato da
Mirla Conteiro Ruiz DiazCopyright
Formati disponibili
Condividi questo documento
Condividi o incorpora il documento
Hai trovato utile questo documento?
Questo contenuto è inappropriato?
Segnala questo documentoCopyright:
Formati disponibili
Voltametria e Polarografia
Caricato da
Mirla Conteiro Ruiz DiazCopyright:
Formati disponibili
UNIDADE
VOLTAMETRIA/POLAROGRAFIA: CONCEITOS E TCNICAS
Mtodos Eletroanalt!"os
Os mtodos eletroanalticos so mtodos instrumentais de anlises que empregam as
propriedades eletroquimicas de uma soluo para determinas a concentrao de um analito.
Anal!to
Em qumica analtica, analito o componente (elemento, composto ou on) de interesse
analtico de uma amostra.
A informao analtica que se obtm sobre o analito na amostra pode ser qualitativa (se
o analito est presente ou numa determinada concentrao na amostra), quantitativa (a
proporo em que se encontra) e estrutural.
A voltametria uma tcnica eletroqumica onde as informaes qualitativas e
quantitativas de uma espcie qumica so obtidas a partir do registro de curvas corrente-
potencial, feitas durante a eletrlise dessa espcie em uma cela eletroqumica constituda de
pelo menos dois eletrodos, sendo um deles um microeletrodo (o eletrodo de trabalho) e o outro
um eletrodo de superfcie relativamente grande (usualmente um eletrodo de referncia). O
potencial aplicado entre os dois eletrodos em forma de varredura, isto , variando-o a uma
velocidade constante em funo do tempo. O potencial e a corrente resultante so registrados
simultaneamente. A curva corrente vs. potencial obtida chamada de voltamograma.
Na voltametria, o potencial aplicado a um eletrodo o parmetro de controle e variado
de forma sistemtica de modo a produzir uma reao redox sobre o eletrodo. A corrente, por
outro lado, resultante da transferncia de eltrons que ocorre durante a reduo ou a oxidao
de espcies eletroativas; sobre a superfcie do eletrodo.
Entre o final do ano 1950 e o incio de 1960, o desenvolvimento de tcnicas
espectroscpicas diminuiu consideravelmente a utilizao da polarografia em anlises, exceto
em aplicaes especiais, como a determinao de oxignio molecular em solues. A partir da
metade dos anos 60, com o desenvolvimento de amplificadores operacionais rpidos e estveis,
importantes modificaes na polarografia clssica foram desenvolvidas, de modo a aumentar
significantemente a sensibilidade e a seletividade do mtodo. Atualmente, instrumentos
relativamente baratos e com sensibilidade, em parte por bilho para muitas substncias
eletroativas so comercializados. Desta forma, um recente ressurgimento no interesse da
utilizao da polarografia tem sido comprovado.
Como a rea dos dois eletrodos diferente, o microeletrodo se polarizar, isto ,
assumir o potencial aplicado a ele. O eletrodo de referncia, por possuir uma rea grande, no
se polarizar, mantendo o seu potencial constante. O microeletrodo comumente feito de um
material inerte, como Au, Pt, Hg e C. Quando o microeletrodo constitudo de um eletrodo
gotejante de mercrio, a tcnica chamada de polarografia.
Os microeletrodos tm formatos e tamanhos diferentes, com frequncia so discos
pequenos de um condutor que prensado em um cilindro de material inerte, como Teflon, que
tenha em si, um fio para contato (Figura 1). O condutor pode ser um material inerte, como Pt,
Au. Em voltametria os sinais de excitao (Figura 1), de potencial varivel, so aplicados sobre
uma clula eletroqumica contendo um microeletrodo. Esse sinal extrai uma resposta
caracterstica de corrente na qual se baseia o mtodo.
1
FIGURA #: Microeletrodos FIGURA #$: Sinais de excitao
Nas ltimas dcadas, o desenvolvimento de novos circuitos eletrnicos tem permitido
desenvolver instrumentos com capacidade de aplicar diferentes formas de programao de
potencial e, dependendo da programao estabelecida, a tcnica polarogrfica recebe distintos
nomes, como ser visto, posteriormente.
A instrumentao bsica da polarografia consiste de trs partes principais: um
potenciostato, um gerador de funes e um microampermetro.
A funo do potenciostato aplicar uma voltagem entre o eletrodo de trabalho e o
eletrodo auxiliar, ou seja, manter uma diferena de potencial constante entre o eletrodo de
trabalho e o eletrodo de referncia.
O gerador de funes deve variar este potencial, tanto na direo negativa ou positiva,
de forma contnua ou em saltos entre dois valores distintos de potencial.
O microampermetro deve registrar a corrente que circula na clula eletroqumica de
modo sincronizado corri a variao do potencial.
A corrente devida transferncia de eltrons que se processa quando oxidao (perda
de eltrons) ou reduo (ganho de eltrons) ocorre sobre a superfcie do eletrodo denominada
de corrente faradica. Esta corrente proporcional concentrao das espcies eletroativas em
soluo. A corrente de reduo (corrente catdica) , por conveno, assinalada com um sinal
positivo, enquanto que a corrente de oxidao (corrente andica) com um sinal negativo.
2
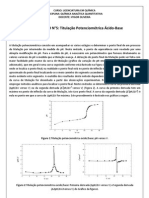
Os primeiros estudos voltamtricos foram feitos por Heyrovsky e Kuceras, em 1922,
usando um eletrodo gotejante de mercrio como eletrodo de trabalho e como eletrodo de
referncia, o ECS. Portanto, a primeira tcnica voltamtrica desenvolvida foi a polarografia. A
curva corrente vs voltagem obtida, chamada de polarograma. A Figura 2, mostra um
polarograma obtido para uma soluo de Cd(II) em HCl 1 mol/L.
FIGURA %: Polarogramas de corrente contnua (DC) tpicos: (A) polarograma de 0,5x10
-3
mol/L
Cd
+2
em HCl 1,0 mol/L. (B) Polarograma de HCl 1,0 mol/L.
Para melhor entendimento, o polarograma mostrado na Figura 2 pode ser dividido em
cinco partes:
1. Regio onde o potencial positivo (E > 0): surge uma corrente andica devido a oxidao do
mercrio do prprio eletrodo de trabalho:
Hg Hg
2+
+ 2e
-
portanto, nessa regio a polarografia no pode ser usada.
2. Regio entre 0 V e -0,5 V (0 V < E < -0,5V): nessa parte do polarograma observa-se apenas
a chamada corrente residual, que decorrente de reduo/oxidao de impurezas presentes no
eletrlito de suporte (HCl 1mol L
-1
).
3. Parte do polarograma onde E = -0,6 V: Neste potencial ocorre um aumento brusco da
corrente em funo da reduo do cdmio junto superfcie do eletrodo gotejante de mercrio
Cd
2+
+ 2e
-
+ Hg Cd(Hg)
4. Regio de -0,7 V < E < -1V: Nessa parte do polarograma a corrente atinge um valor limite e
por isso chamada de corrente limite e independente do potencial aplicado. Nesse intervalo de
potencial o Cd
2+
reduzido to rapidamente quanto chega na superfcie do eletrodo, atravs de
um processo de transporte por difuso de seus ons do interior da soluo at superfcie do
eletrodo. Como a soluo mantida sem agitao, o transporte de massa da espcie eletroativa
(Cd
2+
) no envolver conveco. Como a soluo tambm possui um eletrlito de suporte (HCl 1
3
mol/L), o transporte de massa da espcie eletroativa no envolver migrao, o que produziria
uma corrente de migrao devido movimentao de espcies carregadas sob efeito de um
campo eltrico.
Tendo HCl 1 mol/L como eletrlito de suporte, a corrente de migrao ser praticamente
devido ao HCl. Assim, este transporte do Cd
2+
do seio da soluo junto superfcie do eletrodo
ser governado apenas por um processo difusional. Nessa condio, a corrente resultante
chamada de corrente de difuso. Como pode ser visto na Figura 2, essa corrente obtida pela
diferena entre a corrente residual e a corrente limite, e representada por id.
A relao entre a corrente de difuso (que uma corrente do tipo faradaica, isto , uma
corrente produzida por uma reao eletrdica) e a concentrao da espcie eletroativa em
soluo dada pela equao de Ilkovic:
onde
id = corrente de difuso (A)
n = quantidade de matria (antigamente conhecido como "nmero de moles) de
eltrons por mol de substncia
m = velocidade da vazo de mercrio atravs do capilar de vidro (mg/s)
t = tempo de gota (s)
C = concentrao em mmol L
-1
.
Alm da difuso, como comentado acima, mais dois processos de transferncia de massa
entre a soluo e a superfcie do eletrodo podem ocorrer. Um desses processos a migrao de
partculas carregadas em um campo eltrico. O outro a conveco, um processo mecnico, que
ocorre devido movimentao da soluo (usando-se um agitador magntico e uma barra
magntica, por exemplo). O processo de migrao em um campo eltrico minimizado pela
adio de um eletrlito inerte (eletrlito de suporte) soluo em uma concentrao pelo menos
100 vezes maior do que a substncia eletroativa (HCl 1 mol/L, neste caso). O processo de
conveco eliminado mantendo-se a soluo em repouso, sem agitao. Assim, apenas o
processo de difuso ser responsvel pelo transporte de massa, e a corrente medida, id, pode
ser efetivamente expressa como corrente de difuso.
5. Regio do polarograma onde E < -1,0 V: A corrente aumenta em funo do potencial devido
reduo de H3O
+
(simplificadamente, H
+
) do eletrlito de suporte:
H
+
+ e
-
H2
Nessa regio a polarografia tambm no pode ser usada, pois a corrente devido ao
eletrlito de suporte (ons H
+
, neste exemplo) sobrepor-se- corrente de difuso produzida
pela espcie eletroativa de interesse (analito).
O potencial no polarograma correspondente meia altura da onda polarogrfica (no
ponto onde i = id/2), cujo valor est ao redor de -0,6 V conforme pode ser visto no exemplo da
Figura 1, chamado de potencial de meia-onda, representado por E1/2. O potencial de meia onda
caracterstico da substncia eletroativa e reflete a facilidade de reduo ou oxidao da
substncia em um dado eletrlito. O valor de E1/2, desse modo, serve para identificar a espcie
eletroativa, ou seja, para fazer a anlise qualitativa de espcies presentes em uma dada
amostra.
Do ponto de vista de anlise quantitativa a informao importante do polarograma o
fornecimento do valor da corrente de difuso, id. A corrente de difuso relacionada
concentrao da substncia eletroativa pela equao de Ikovic, conforme discutido acima no
tem 4, equao 1. Esta equao pode ser simplificada para:
4
onde k envolve os termos (607nD
1/2
m
2/3
t
1/2
) da eq. 1, uma vez que eles permanecem constantes
em um dado eletrlito de suporte, para um mesmo capilar, uma mesma temperatura, e uma
mesma presso de Hg sobre o capilar. A eq. 2 chamada de equao simplificada de Ilkovic
que, por ser mais conveniente do que a equao 1, preferencialmente usada para fins
analticos.
Quanto ao potencial de meia-onda, E1/2, trata-se de um parmetro oriundo da equao
de Nernst aplicada polarografia. Esta equao considera que a reao eletroqumica junto
superfcie do eletrodo gotejante de mercrio reversvel. De um modo geral, a reao
representada por:
Ox + ne
-
Red
A equao de Nernst para esta reao no sentido da reduo (onda polarogrfica
catdica), a 250 C, adquire a forma:
onde :
E = potencial devido relao de concentraes na interface eletrodo/soluo das
formas oxidada e reduzida da espcie eletroativa ([Ox]i/[Red]),
E
0
= potencial padro do sistema de xido reduo constitudo pelas formas oxidada e
reduzida da espcie eletroativa,
[Ox]
i
= concentrao da forma oxidada da espcie eletroativa junto interface eletrodo-
soluo,
[Red]i = concentrao da forma reduzida da espcie eletroativa junto interface
eletrodo-soluo.
A partir desta considerao inicial, e das consideraes sobre a relao entre as
correntes e concentraes das formas oxidada e reduzida da espcie eletroativa, expressas pela
equao de Ilkovic (eq. 2) tem-se:
sendo:
i = corrente em qualquer ponto da onda polarogrfica,
[Ox] = concentrao da forma oxidada da espcie eletroativa no seio da soluo e
K = constante da equao de Ilkovic para a forma oxidada da espcie eletroativa.
No plat da onda polarogrfica, [Ox]
i
, a concentrao da forma oxidada da espcie
eletroativa na interface do eletrodo, torna-se zero, pois toda partcula da espcie eletroativa
que chegar junto superfcie do eletrodo (governada por difuso, como a
polarografia/voltametria) ser reduzida, e a corrente ser = corrente de difuso. A eq. (4) se
torna:
5
onde id a corrente de difuso. Combinando-se as equaes (4) e (5) chega-se a:
Se a forma reduzida da espcie eletroativa for solvel em gua e esta forma no estiver
presente originalmente junto com a forma oxidada, ela se difundir da superfcie do eletrodo
para o corpo da soluo, ou, no caso de metais, elpoder se difundir da superfcie do eletrodo
para o interior da gota de mercrio, formando amlgama. Assim, para qualquer valor de:
onde k a constante da equao de Ilkovic para a forma reduzida da espcie eletroativa. Aqui a
corrente i depender apenas da concentrao da forma reduzida junto superfcie do eletrodo,
uma vez que a concentrao da forma reduzida originalmente presente igual a zero.
Substituindo-se na equao (3), chega-se a
ou
onde
Quando a corrente i for igual metade da corrente de difuso ( i =i d/2), o potencial ser
igual ao potencial de meia onda, E1/2, ( Figura 1)e a equao (9) se reduz :
Desta equao pode-se ver que o potencial de meia onda, E1/2, constante e
caracterstico para uma dada substncia eletroativa que constitua um sistema de xido-reduo
reversvel e que seu valor independente da concentrao da forma oxidada, [Ox], no corpo da
soluo. Assim, combinando-se as equaes (9) e (11) , chega-se :
Esta equao chamada de equao da onda polarogrfica e representa o potencial como uma
funo da corrente em qualquer ponto da onda polarogrfica.
6
O potencial de meia onda caracterstico da substncia eletroativa, no dependendo nem
mesmo das caractersticas do eletrodo. Por isso ele til na anlise qualitativa de amostras
desconhecidas, podendo ser usado na identificao de substncias presentes nessas amostras.
Procedendo-se maneira anloga para um processo andico, teremos a seguinte equao
para uma onda andica:
AS CLULAS VOLTAMTRICAS
As clulas eletroqumicas utilizadas em voltametria/polarografia so, evidentemente , do
tipo eletroltica e podem ter dois ou trs eletrodos. Heyrovsky utilizou uma clula de dois
eletrodos e durante muito tempo usou-se apenas esse tipo de clula. Na clula de dois eletrodos
(Figura 2), conforme j foi exposto na introduo, tem-se um eletrodo de trabalho, de superfcie
pequena, ou seja, um microeletrodo. No caso da polarografia o eletrodo de trabalho um
microeletrodo gotejante de mercrio. O potencial aplicado no eletrodo de trabalho frente a um
eletrodo de referncia, usualmente um eletrodo de calomelano saturado, de rea superficial
grande, para que apenas o eletrodo de trabalho polarize. Essa clula apresenta alguns
inconvenientes, pois a corrente resultante da varredura de potencial passa atravs do eletrodo
de referncia. Isto obriga a usar-se um eletrodo de calomelano de reservatrio grande de KCl,
devido reao que ocorrer no mesmo.
Por exemplo: na determinao polarogrfica de cobre, haver reduo de Cu(II) no
eletrodo de mercrio (eletrodo de trabalho), e, conseqentemente oxidao de Hg na outra meia
clula (calomelano saturado). Assim, na meia clula do eletrodo de trabalho:
Cu
+2
+ 2 e
-
Cu
Na meia clula do calomelano:
Ou seja, haver consumo de Hg0 e Cl
-
, necessitando usar-se eletrodos de referncia grandes,
para que eles mantenham o potencial constante durante a aplicao de potencial.
Devido passagem de corrente atravs do eletrodo de referncia e reaes que ocorrem
no mesmo, isto afetar as medidas em concentraes da ordem de 10
-4
mol L
-1
, pois essa
corrente se aproximar do valor da corrente de difuso. Outra limitao a resistncia da clula.
Quando ela aumenta, como no caso de meio no aquoso, aumentar a corrente que passar
atravs dos eletrodos o que provocar distores nos polarogramas, tornando invivel a
utilizao da tcnica nessas condies.
7
CLULAS DE TR&S ELETRODOS
Para resolver essas limitaes das clulas de dois eletrodos, foi desenvolvida a clula de
trs eletrodos (Figura 3a). O terceiro eletrodo chamado de eletrodo auxiliar, podendo ser de
platina, ouro, carbono vtreo, etc. Ele foi introduzido na clula voltamtrica para assegurar o
sistema potenciosttico. Nesta clula, os eletrodos so conectados a um amplificador
operacional, pertencente ao circuito eletrnico do polargrafo. O amplificador operacional atuar
quando for aplicada uma diferena de potencial entre o eletrodo de trabalho e o eletrodo de
referncia, fazendo com que a resistncia do eletrodo de referncia aumente e a do eletrodo
auxiliar diminua (Figura 3b). Assim, a corrente passar entre o eletrodo de trabalho e o auxiliar,
evitando que ocorram distrbios (como eletrlise, por exemplo) no eletrodo de referncia. Com
este recurso o eletrodo de referncia realizar o seu papel sem interferncias, que o de manter
o seu potencial constante durante as medidas. Por isto pode-se usar alm do eletrodo de
trabalho e do auxiliar, um eletrodo de referncia de dimenses pequenas, o que facilita o uso de
recipientes polarogrficos/voltamtricos de tamanho reduzido.
De um modo geral, a clula de trs eletrodos apresenta as vantagens de:
1. ser mais adequada para solues diludas,
2. poder ser usada para solues de alta resistncia (solventes orgnicos, mistura gua mais
solvente orgnico),
3. poder ser usada com eletrlitos de suporte mais diludos.
CLULA ELETRO'U(MICA E INSTRUMENTA)*O
Os experimentos eletroqumicos usualmente so realizados em uma clula contendo trs
eletrodos imersos na soluo a ser analisada (vide Figura 3):
1) O eletrodo de trabalho, onde se processa a reao de interesse. Na polarografia
clssica, o eletrodo de trabalho utilizado o eletrodo gotejante de mercrio;
2) O eletrodo de referncia, que fornece um potencial estvel, com o qual o potencial
aplicado ao eletrodo de trabalho comparado. Os eletrodos de referncia mais utilizados em
polarografia so o eletrodo de calomelano saturado (ECS) e o eletrodo de prata/cloreto de prata
(Ag/AgCl);
3) O eletrodo auxiliar (ou contra eletrodo), que consiste de um material condutor e
quimicamente inerte, como por exemplo, platina (Pt).
FIGURA + - Clula de trs eletrodos, utilizada em experimentos eletroqumicos
8
A soluo a ser analisada deve conter a amostra dissolvida e um eletrlito de suporte
tambm dissolvido, numa concentrao mnima de 10
-3
mol/L, para assegurar uma
condutividade mdia da soluo requerida nas medidas eletroqumicas.
O eletrlito de suporte deve ser escolhido de forma a no interferir nas reaes
eletroqumicas das espcies eletroativas analisadas no intervalo de potencial de interesse.
Entre os eletrlitos de suporte tpicos podemos destacar os do tipo
cido: (HCI, HNO3, HClO4, H2SO4, etc.);
Bsico: (NaOH, KOH, NH4OH, etc.);
Sal: (NaCI, KCI, K2SO4, etc.);
Tampo: (acetato, fosfato citrato, tartarato, etc.).
ELETRODO GOTE,ANTE DE MERC-RIO
Quanto ao eletrodo gotejante de mercrio, pelo fato de ser um eletrodo lquido,
constitudo por um reservatrio de mercrio conectado a um tubo capilar de vidro, com
comprimento variando entre 5 e 20 cm (vide Figura 4). O mercrio, forado pela gravidade,
passa atravs desse tubo, com cerca de 0,02 a 0,05 mm de dimetro interno, formando um
fluxo constante de gotas idnticas, cujos dimetros podem variar de 0,2 a 1 mm.
9
FIGURA .: Clula polarogrfica/voltamtrica de trs eletrodos. (a) Esquema mostrando os
eletrodos no recipiente polarogrfico (b) Esquema com a clula conectada a um
potenciostato.
As gotas se formam em intervalos de tempo definidos, entre 1 e 5 segundos, devido
presso constante exercida pelo mercrio (Figura 4a). O capilar de vidro ligado ao reservatrio
por um tubo plstico flexvel. O conjunto capilar mais tubo plstico mais reservatrio chamado
de coluna de mercrio. O tempo de durao da gota controlado pela gravidade variando-se a
altura dessa coluna, o que facilmente conseguido porque o tubo plstico flexvel. Este tipo de
meia-clula de mercrio nasceu com a polarografia e utilizado nos instrumentos de clulas
com dois eletrodos (Figura 4b).
10
Nos polargrafos modernos o gotejamento do mercrio passa a ser controlado por
dispositivos eletromecnicos, devido aos recursos instrumentais incorporados nos aparelhos
modernos. Uma vlvula solenide abre e fecha o duto do mercrio, sincronizada a um dispositivo
(chamado de martelo) que golpeia o capilar, derrubando a gota. Isto permite ao usurio
controlar o tempo de durao e o tamanho da gota selecionando-os no painel do instrumento em
uso.
Escolhe-se um tamanho e um tempo pr-determinado para a durao da gota de
mercrio, a vlvula solenide abre e fecha rapidamente aps a gota atingir o tamanho escolhido,
o martelo acionado a seguir no tempo pr-determinado golpeando o capilar e derrubando a
gota. Logo uma nova gota formada, repetindo-se o ciclo sucessivamente at ao final da
varredura de potencial.
Todo o conjunto de operaes, envolvendo formao da gota, tempo de durao da
gota, varredura de potencial, medida da corrente e registro do polarograma/voltamograma
feito de maneira sincronizada e automtica, em razo dos recursos eletrnicos presentes nos
polargrafos. Esta meia-clula de mercrio a preferida para ser usada em sistemas de clulas
de trs eletrodos.
O M/0IMO POLAROGR/FICO
O mximo polarogrfico um fenmeno que ocorre durante o registro do polarograma
devido a turbulncias envolvendo a gota de mercrio e a camada de difuso adjacente a ela. No
incio do plat da onda polarogrfica a corrente de difuso fica maior do que deveria, voltando
aps mais alguns milivolts de varredura ao nvel normal, governada pelo processo de difuso.
Esse mximo interfere na anlise polarogrfica pela distoro da forma da onda, o que dificulta a
determinao de sua altura (corrente de difuso, id). Na maior parte das vezes ele tem uma
forma aguda, como mostrada na Figura 5, e chamado de mximo de primeira ordem.
FIGURA 1: Polarografia DC de uma soluo de In (III) mostrando um mximo polarogrfico de
primeira ordem.
11
Geralmente esse mximo explicado em termos de uma no uniformidade do campo
eltrico devido geometria da gota de mercrio, ou seja, a densidade de corrente seria
desigualmente distribuda na gota. Ela seria maior na parte da gota que est presa ao capilar do
que no fundo da gota. Isto faria com que a camada deslizasse na superfcie da gota de mercrio
aumentando momentaneamente a quantidade de partculas que reagiriam no eletrodo e
conseqentemente a corrente medida nessa parte do polarograma. Aps essa quantidade de
partculas extras reagirem, ela se reduziria quantidade governada pela difuso e a corrente se
normalizaria.
A maneira de evitar a formao de mximos na prtica utilizar os chamados
supressores de mximo, que so substncias tenso-ativas. As molculas dessas substncias so
adsorvidas junto superfcie da gota de mercrio formando um filme protetor, o que impediria o
deslizamento da camada de difuso. O supressor mais comum a gelatina, que utilizada a
uma concentrao de 10-3 a 10-4 % (m/V). Outros supressores comuns so o vermelho de
metila e o triton X-100, sempre usados em baixas concentraes para no isolarem a superfcie
do eletrodo devido espessura do filme de adsoro.
Em alguns casos o mximo polarogrfico tem a forma arredondada e ocorre em solues
de eletrlitos concentradas (a partir de 0,1 mol/L). Esse tipo de mximo chamado de mximo
de segunda ordem e tambm evitado usando-se supressores de mximo como no caso
anterior. A causa para esse mximo atribuda a processos convectivos ocorrendo dentro da
gota de mercrio (movimentao do mercrio dentro da prpria gota) mais que na soluo.
Fenmenos de adsoro tambm podem produzir mximos polarogrficos. Nesse caso o
mximo chamado mximo de terceira ordem. Ele ocorre em razo da adsoro de substncias
que apresentam propriedades superfcie-ativas. Na regio do polarograma em cujo potencial a
adsoro ocorrer haver um aumento da corrente de modo semelhante aos casos anteriores.
ELETRODOS DE TRA2AL3O
O eletrodo de trabalho mais importante usado em voltametria ainda o eletrodo de
mercrio. Ele pode ser usado tanto no modo gotejante quanto no modo esttico. Nesse modo, a
gota de mercrio, depois de formada, permanece imvel na ponta do capilar e o voltamograma
registrado nessa nica gota. O eletrodo de mercrio tambm pode ser usado na forma de filme
de mercrio. Neste caso, o filme depositado eletroquimicamente em superfcies slidas, como
de platina e carbono vtreo. A sua atuao (do eletrodo de mercrio) marcadamente na regio
catdica, em potenciais que podem variar de +0,3 V a -2,3 V vs. o eletrodo de calomelano
saturado (ECS), dependendo do meio utilizado. Esta regio de potencial onde ocorrem as
reaes eletrdicas da maioria dos ons metlicos e de grande nmero de espcies orgnicas, da
a sua importncia. No modo gotejante ele um eletrodo de superfcie renovvel, evitando
problemas, entre outros, devido aos chamados envenenamentos superficiais.
O eletrodo gotejante de mercrio (DME) foi inventado por Heyrovsky para medidas de
tenso superficial e representa, historicamente e atualmente, um instrumento de enorme
importncia para a eletroanaltica. A Figura 4 mostra o diagrama de um DME. O mercrio est
contido em um reservatrio e liberado atravs de um fino tubo capilar (dimetro interno de
-0,05 mm) na forma de gotas esfricas. Cada gota cresce at que seu peso no possa mais ser
suportado pela tenso superficial. Tipicamente, o tempo de vida de uma gota de 2 a 6 s e cada
gota "madura" tem um dimetro de 0,5 a 1,0 mm.
Para um processo de eletrlise ocorrendo durante o crescimento de uma gota de
mercrio, a corrente deve possuir uma dependncia com o tempo, que reflete tanto a expanso
do "eletrodo esfrico" quanto os efeitos da eletrlise. Alm disso, cada gota que se desprende do
capilar agita a soluo e praticamente anula os efeitos de depleo, tanto que cada gota pode
ser considerada como "nascendo em uma soluo fresca e o tempo de vida representando um
novo experimento.
Entre os eletrodos slidos o de platina um dos mais importantes. Ele atua na regio de
potencial de cerca de +1,1 V a -0,5 V vs. ECS, dependendo do eletrlito de suporte utilizado.
12
til na regio andica, onde o eletrodo de mercrio no atua. Outro eletrodo slido til na regio
andica o de ouro, que pode ser utilizado em intervalos de potencial de +1,5 a -0,8 V vs. ECS.
Muito usados na regio andica so os eletrodos de carbono, principalmente o de carbono vtreo,
que atua no intervalo de +1,5 V a -1,1 V vs. ECS, dependendo do meio utilizado.
Embora diversos eletrodos slidos possam ser usados em parte da regio catdica, todos
tm desempenho inferior ao eletrodo de mercrio, devido principalmente as suas propriedades
envolvendo a renovao superficial e formao de amlgamas com vrios metais.
Mais recentemente tm sido desenvolvidos novos tipos de eletrodos de trabalho para
serem usados em voltametria, tais como os eletrodos quimicamente modificados e os ultra-
microeletrodos. Embora em boa parte ainda estejam em estgio de pesquisa, apresentam um
grande potencial para ampliar o campo de utilizao da tcnica voltamtrica.
Na Tabela 1 so apresentados os valores para a reprodutibilidade do tamanho da gota e
na 2, os valores tpicos das reas de gotas de mercrio do sistema Modelo 303 SMDE,
comercializado pela empresa EG&G Pinceton Applied Research Corporation.
TA2ELA # - REPRODUTIBILIDADE DO TAMANHO DA GOTA DE MERCRIO COM O SISTEMA 303
SMDE DA EG&G PARC.
Modo de operao Tamanho da Gota Desvio Padro Relativo (%)
EGM Pequeno 0,79
EGPM Pequeno 0,56
EGPM Grande 0,87
TA2ELA % - reas tpicas da gota de mercrio com o sistema 303 SMDE da EG&G PARC
Tamanho da Gota
Peso da Gota
(mg)
rea da Gota
(cm
2
)
Pequeno 1,2 0,096
Mdio 2,5 0,0156
Grande 5,4 0,0261
O eletrodo gotejante de mercrio possui caractersticas que permitem destacar a
polarografia entre os mtodos eletroanalticos. Entre as vantagens podemos mencionar:
1) O processo de gotejamento faz com que o eletrodo seja sempre renovado, ou seja, a
superfcie do eletrodo no sofre modificaes permanentes por reaes que possam conduzir a
uma deposio de materiais indesejveis. Com qualquer outro eletrodo, a condio da superfcie
do eletrodo dever ser constantemente monitorada. Se o comportamento do eletrodo sofre
alterao por reaes ocorrendo sobre a superfcie, procedimentos devem ser adotados para
regenerar a superfcie original.
2) O alto sobrepotencial da descarga de hidrognio sobre mercrio e a habilidade deste
em formar amlgama com muitos ons metlicos, permite que a anlise possa ser conduzida em
13
potenciais mais negativos que qualquer outro eletrodo metlico e toma possvel o estudo de
reaes que so termodinamicamente menos favorveis.
Um exemplo a reduo do on sdio a amlgama de sdio em soluo aquosa alcalina,
que pode ser claramente observada em um experimento polarogrfico.
A grande desvantagem do eletrodo gotejante de mercrio o seu limite andico. A
oxidao do mercrio cm solues aquosas cidas, por exemplo, ocorre em potenciais prximos
a 0,0 V vs. ECS e no permite a anlise de materiais que so oxidados em potenciais mais
positivos.
REMO)*O DO O0IG&NIO DISSOLVIDO
Quando se trabalha na regio catdica, como o caso da polarografia, h a necessidade
da remoo do oxignio atmosfrico dissolvido nas solues. Isto porque o O2 eletroativo e
produz duas ondas polarogrficas nessa regio, uma com potencial de meia onda, E1/2, ao redor
de -0,05 V vs. o eletrodo de calomelano saturado (ECS) e a outra com E1/2 ao redor de -1,0 V vs.
ECS. A primeira onda catdica devido s reaes:
O2 + 2 H
+
+ 2e
-
.. H2O2 (meio cido)
O2 + 2 H
+
+ 2e
-
.. 2 H2O (meio alcalino ou neutro)
Tanto a formao de gua oxigenada na reduo do oxignio em meio cido como a formao
de gua em meio alcalino ou neutro ocorrero no mesmo potencial (E1/2 -0,05 V vs. ECS).
A segunda onda catdica (E1/
2
-1,0 V vs. ECS) devido s reaes:
O2 + 4 H
+
+ 2e
-
H2O2 (meio cido)
O2 + 2 H2O + 4e
-
4 OH
-
(meio alcalino ou neutro)
Como consequncia, na regio catdica quando se faz uma medida polarogrfica ou voltamtrica
na presena de O2 a corrente de fundo ser alta, e vai mascarar a corrente produzida pela
espcie eletroativa. Por isso necessrio remover o O2 dissolvido na soluo antes das medidas
serem feitas. Isto feito desaerando-se a soluo, pela passagem de um gs inerte, isento de
O2. O gs borbulhado na soluo durante alguns minutos, remove o O2, e fica dissolvido em
seu lugar. Mas como eletroquimicamente inerte nesse intervalo de potencial (regio catdica
considerada acima) no produzir nenhuma corrente polarogrfica ou voltamtrica. Os gases
mais usados para esse fim so: N2, Ar, Ne e He.
O Nitrognio o mais usado por ser mais barato e poder ser facilmente obtido com pureza
alta em relao presena do oxignio. Quando isto no ocorre, podem ser usados frascos
lavadores de gs intercalados entre o cilindro de N2 e o recipiente polarogrfico, contendo
redutores que removero o O2, purificando assim o nitrognio que ser borbulhado na soluo a
ser polarografada.
FATORES 'UE AFETAM A VELOCIDADE DE UMA REA)AO
NA INTERFACE ELETRODO4SOLU)*O
Consideremos uma reao composta de diversas etapas, ocorrendo na interface
eletrodo-soluo (reao eletrdica), e produzindo a oxidao de espcies dissolvidas, O, em
espcies reduzidas, R, tambm em soluo (Figura 6), ou seja, a reao global pode ser escrita
como:
O + ne
-
R
14
Em geral, a corrente governada pelas velocidades dos seguintes processos:
1) Transferncia de massa do seio da soluo para a superfcie do eletrodo;
2) Transferncia de cargas na superfcie do eletrodo;
3) Reaes qumicas precedentes ou subsequentes transferncia de carga. Estas
podem ser processos homogneos (em soluo), como protonao ou dimerizao,
ou heterogneos (na superfcie do eletrodo), como decomposio cataltica;
4) Outras reaes superficiais, como adsoro, dessoro, ou cristalizao
(eletrodeposio).
F!56ra 7 - Etapas de uma reao eletrdica geral
REA)*O ELETR8DICA REVERS(VEL OU NERNSTIANA
As reaes eletrdicas mais simples so aquelas cujas cinticas de transferncia de
cargas e etapas qumicas associadas so muito rpidas quando comparadas com os processos de
transporte de massa. Em outras palavras, se um processo eletrdico envolve somente etapas
com cinticas de transferncia de cargas rpidas e reaes homogneas reversveis, podemos
estabelecer que:
a) as reaes homogneas esto cm equilbrio
b) as concentraes superficiais das espcies envolvidas na reao O + ne
-
R esto
relacionadas ao potencial do eletrodo E por uma equao da forma de Nernst, ou seja:
=
=
+ =
) 0 x ( C
) 0 x ( C
ln
nF
RT
E E
R
0 0
,
onde E
o
o potencial padro, x a distncia da superfcie do eletrodo, Co(x = O) e CR(x = O) as
concentraes das espcies O e R na superfcie do eletrodo, e os demais termos com seus
significados usuais. Estas reaes eletrdicas so freqentemente chamadas de reaes
reversveis ou reaes nernstianas.
Assim, a velocidade "lquida", v, de uma reao nernstiana totalmente governada pela
velocidade com que as espcies eletroativas so trazidas para a superfcie do eletrodo por
transporte de massa, vtm. Temos ainda que:
15
nFA
i
v v
tm
= =
Podemos notar que o valor de I denominado de constante da corrente de difuso,
depende somente de n e Do, que so constantes caractersticas das espcies eletroativas e da
soluo. Esta constante I pode ser entendida da mesma forma como a absortividade molar, ,
uma constante do sistema para medidas ticas. O produto (m
2/3
t
1/6
), chamado de constante do
capilar, descreve a influncia das caractersticas do eletrodo gotejante sobre a corrente de
difuso; ambos m e t so fceis de serem avaliados experimentalmente. Portanto, conhecendo-
se I, podemos avaliar Co* simplesmente pelas medidas de id.
TRANSPORTE DE MASSA EM CLULAS ELETRO'U(MICAS
O transporte de massa, isto , o movimento de espcies de um local na soluo para
outro, surge das diferenas no potencial eltrico ou qumico nos dois locais, ou do movimento de
um elemento de volume na soluo. Os modos de transporte de massa so:
1. Migrao: movimento de um corpo carregado sob a influncia de um campo eltrico (um
gradiente de potencial eltrico);
2. Difuso: movimento de espcies sob a influncia de um gradiente de potencial qumico,
ou seja, um. gradiente de concentrao;
3. Conveco: transporte hidrodinmico ou por agitao. Geralmente, o fluxo de um fluido
ocorre por conveco natural (conveco causada por gradientes de densidade) e
conveco forada, e pode ser caracterizado por regies estagnantes, fluxo laminar e
fluxo turbulento.
O transporte de massa para um eletrodo governado pela equao de Nernst-PIanck, a
qual para transporte unidimensional ao longo do eixo x pode ser escrita como
) x ( v C
) x (
) x ( C
D
) x (
) x (
C D
RT
z
(x) J
i
i
i i i
i
i
+
=
,
onde:
Jj(x) o fluxo de espcies i (mol s
-1
cm
-2
) a uma distncia x da superfcie;
Di o coeficiente de difuso (cm
2
s
-1
);
) x (
) x ( C
i
o gradiente de concentrao a uma distncia x;
) x (
) x (
o gradiente de potencial;
zi e Ci a carga e a concentrao das espcies i, respectivamente;
Vi(x) a velocidade (em s
-1
) com a qual um elemento de volume na soluo se move ao longo do
eixo x.
Nesta equao, os trs termos do lado direito representam as contribuies da migrao,
difuso e conveco, respectivamente, ao fluxo.
TRATAMENTO SEMI4EMP(RICO PARA UMA REA)*O ELETR8DICA
SO2 CONTROLE DIFUSIONAL
Transporte de massa por migrao no apresenta utilidade em experimentos
eletroqumicos. Na presena de um excesso de eletrlito de suporte na soluo, a migrao das
espcies eletroativas minimizada a uma extenso tal que pode ser desprezada. Por outro lado,
desde que um gradiente de concentrao sempre se desenvolve to logo uma reao eletrdica
16
seja iniciada, transporte de massa por difuso desempenha um papel fundamental em qualquer
experimento eletroqumico.
Assim sendo, consideremos um eletrodo planar, de rea A, imerso em uma soluo
inicialmente contendo somente espcies O que so reduzidas de acordo com a reao:
O + ne
-
R
j aqui demonstrada.
A soluo contm um excesso de eletrlito de suporte e a clula eletroqumica est livre
de distrbios mecnico e trmico, ou seja, migrao e conveco esto ausentes. Alm disso, as
dimenses da clula so grandes quando comparadas com as do eletrodo. Sob tais condies,
possvel supor que o transporte de massa ocorre pelo processo denominado de difuso linear
semi-infinita.
VOLTAMETRIA 9POLAROGRAFIA: CL/SSICA
Os princpios discutidos at aqui constituem a atualmente chamada
voltametria/polarografia clssica ou de corrente contnua. Do ponto de vista analtico esta
tcnica muito limitada para os dias atuais, pois no consegue determinar espcies com
concentraes abaixo de 10
-4
a 10
-5
mol L
-1
. A atual anlise de traos exige determinao de
concentraes da ordem de at 10
-12
mol L
-1
, ou mesmo menor.
Uma das principais limitaes da polarografia/voltametria clssica em relao
sensibilidade a chamada corrente capacitiva, que uma das principais componentes da
corrente de fundo, que constitui a corrente residual mostrada nos polarogramas da Figuras 2 e
7.
FIGURA ;: Variao da corrente em funo do tempo em voltametria/polarografia. O
valor da corrente faradica (Id) diminui mais lentamente que o da
corrente capacitiva(Ic), permitindo uma medida discriminatria entre
elas.
Essa corrente surge devido a fenmenos relacionados dupla camada eltrica. Quando
um potencial aplicado a um microeletrodo ele adquire uma carga tipo condensador, devido ao
carregamento e descarregamento da dupla camada eltrica. Como em voltametria usa-se
varredura de potencial, a cada etapa de potencial aplicado o eletrodo apresentar esse
carregamento e descarregamento da dupla camada, desenvolvendo uma corrente, que, por esta
razo, chamada de corrente capacitiva ou de condensador.
17
A outra componente da corrente de fundo ou residual uma corrente do tipo faradica,
que ocorre devido a impurezas presentes no eletrlito de suporte, em outros reagentes
utilizados e ao oxignio dissolvido. Essa corrente pode ser reduzida ou mesmo eliminada
usando-se reagentes mais puros e removendo-se o oxignio pela passagem de um gs inerte.
Assim, ficou claro que a principal componente da corrente de fundo que limita a
sensibilidade a corrente capacitiva. Ento a pesquisa cientfica direcionou-se na busca de
procurar reduzir essa corrente para que a sensibilidade da tcnica pudesse ser melhorada.
Portanto, a razo para o baixo limite de deteco da polarografia clssica porque a corrente
faradica, isto , a corrente devido reao no eletrodo da substncia a ser determinada, fica
da mesma ordem ou menor do que a corrente capacitiva quando a sua concentrao atinge
valores da ordem de 10
-4
a 10
-5
mol L
-1
, no sendo mais possvel distingui-la da corrente de
fundo. Desse modo, conseguindo-se discriminar essas correntes, a sensibilidade da tcnica pode
ser melhorada.
Em um experimento de polarografia, o analisador polarogrfico, varre o intervalo de
potencial no qual a reduo (ou oxidao) da espcie eletroativa de interesse ocorre. O mtodo
mais simples de se aplicar uma varredura de potencial na forma de uma "rampa" linear, como
a ilustrada na Figura 8. Esta a programao de potencial utilizada na polarografia DC ("Direct
Current Polarography").
FIGURA < - Programao de potencial para a polarografia DC.
Um primeiro resultado positivo foi obtido na prpria polarografia/voltametria de corrente
contnua, ou, como tambm chamada, polarografia/voltametria DC (do ingls Direct Current),
ou ainda, polarografia/voltametria clssica. Este resultado foi obtido fazendo-se a medida da
corrente perto do final do tempo de vida da gota de mercrio. Como foi verificado que a corrente
capacitiva cai mais rapidamente do que a faradica em funo do tempo, fazendo-se a medida
nos ltimos milisegundos da durao da gota, a corrente faradica ser discriminada da
capacitiva. Esta forma de medir a corrente constitui a polarografia DC ou, como chamada na
Europa, polarografia DC tast (de tasten, do alemo, que quer dizer toque).
Para medir-se a corrente necessrio sincronizar-se a os dispositivos envolvidos na
clula com os envolvidos no potenciostato. Assim, escolhe-se um tempo pr-determinado para a
durao da gota de mercrio, digamos, 1 segundo. A vlvula solenide, comandada a partir do
circuito eletrnico do polargrafo, se abrir e a gota se formar no capilar, atingindo um
tamanho tambm pr-estabelecido, fechando-se ento a vlvula. A gota permanecer esttica
durante 1 s e nos ltimos 15 a 20 ms de durao a corrente ser amostrada, e ao final do tempo
escolhido, o "martelo" golpear o capilar derrubando a gota, formando-se em seguida da mesma
maneira uma nova gota de mercrio, repetindo-se o ciclo de medida, sucessivamente at ao
final da varredura de potencial escolhida.
18
E
E
2
E
1
v = 2 5 mV/s
0 t
A DC amostrada ser registrada em funo do potencial aplicado e produzir um
polarograma como o mostrado na Figura 6b. No h aqui os "dentes de serra" apresentados nos
polarogramas da Figura 2 e da Figura 9a, mas uma linha registrada em "saltos" ou "degraus".
Entretanto, a forma desse novo polarograma a mesma dos polarogramas das Figuras 2 e 9b,
ou seja, em forma de onda. Os saltos ou degraus verificados para o registro da corrente (em um
registrador analgico) se deve convenincia de utilizar-se a rampa de potencial aplicado em
etapas, ou seja, sincronizando-se a velocidade de varredura com o tempo de durao da gota.
Por exemplo, usando-se uma velocidade de varredura de 10 mV/s e um tempo de gota de 1 s,
avana-se o potencial de 10 mV a cada segundo, realizando-se as medidas a cada etapa.
Entretanto, as melhorias obtidas no trouxeram de fato benefcios significativos tcnica
voltamtrica, em termos de sensibilidade.
Rigorosamente falando, consegue-se apenas trabalhar-se com conforto em
concentraes da ordem de 10
-4
a 10
-5
mol L
-1
e melhorar um pouco a resoluo, ou seja, a
diferena de potenciais de meia onda entre duas substncias adjacentes pode ser um pouco
menor que na polarografia DC clssica para que possam ser determinadas simultaneamente,
uma vez que os polarogramas ficam mais bem definidos sem "os dentes de serra" da DC
clssica.
FIGURA =: Comparao de um polarograma obtido com as tcnicas (a) DC e
(b) DC amostrada
19
A VOLTAMETRIA 9POLAROGRAFIA: DE PULSO NORMAL
Na dcada de 50, a polarografia de varredura linear deixou de ser uma tcnica analtica
importante devido ao desenvolvimento de mtodos espectroscpicos com melhores limites de
deteco, mais rpidos e mais convenientes.
Alm disso, na polarografia de corrente amostrada, a corrente medida somente em um
perodo de tempo muito pequeno durante a vida da gota, e toda a corrente faradica que flui
antes do perodo de amostragem no utilizada na medio, pelo contrrio, reduz o fluxo de
espcies eletroativas para a superfcie do eletrodo no tempo da medida real. Estas limitaes
foram largamente superadas a partir da metade dos anos 60, com a introduo das tcnicas
polarogrficas de pulso e a comercializao de eletrodos gotejantes de mercrio esttico.
O eletrodo gotejante de mercrio inicialmente mantido em um potencial de base Eb,
durante um perodo de tempo de vida da gota. Neste potencial, a reao em estudo no se
processa, ou seja, corrente faradica no flui no sistema. Quando restarem apenas cerca de 60
milisegundos (ms) do tempo de vida da gota, ou seja, num tempo , o potencial
instantaneamente mudado para um novo valor E. Durante os ltimos 17 ms deste pulso, ou
seja, a partir de um tempo , a corrente medida e registrada em funo do potencial, e a
aplicao do pulso de potencial termina com o retorno do potencial ao valor inicial Eb. Para cada
nova gota de mercrio, o potencial E maior que o da gota anterior. A Figura 10 mostra os
polarogramas obtidos, de pulso normal e de corrente amostrada, para uma soluo 10
-5
mol/L
Cd
2+
em 0,01 mol/L HCl, e comprova as maiores magnitudes das correntes faradicas com o
mtodo de pulso. Note que as escalas de corrente so diferentes.
20
F!56ra #> - Polarograma de uma soluo soluo 10
-5
mol/L Cd
2+
em 0,01 mol/L HCl. (a)
polarografia de pulso normal e (b) polarografia de corrente amostrada.
A VOLTAMETRIA 9POLAROGRAFIA: DE PULSO DIFERENCIAL
Uma melhoria instrumental considervel na discriminao da corrente faradica da
capacitiva viria a ser conquistada com o desenvolvimento das tcnicas de pulso, principalmente
a de pulso diferencial. Neste caso a instrumentao foi desenvolvida de tal modo que as medidas
de corrente e aplicaes de potencial e pulsos de potencial sejam realizados em intervalos de
tempo muito pequenos.
Na polarografia de pulso diferencial a programao de potencial feita aplicando-se um
pulso de potencial superposto em uma rampa de potencial linearmente crescente em
instrumentos analgicos (uma rampa DC) e cada etapa de aplicao do pulso definida pela
varredura de potencial e pelo tempo de gota utilizados. O pulso aplicado de pequena amplitude
(10 a 100 mV) e imposto durante 50 a 60 ms perto do final da vida da gota, quando o
crescimento da gota de mercrio j cessou Figura 11a.
A corrente amostrada em dois intervalos de tempo de cerca de 15 ms cada um; o
primeiro intervalo imediatamente antes da aplicao do pulso (S1) e o segundo prximo do final
do tempo de vida da gota (S2). O valor final da corrente a diferena entre esses dois valores
medidos.
21
FIGURA ##: Representao esquemtica da aplicao do potencial em funo do tempo
em polarografia de pulso diferencial: (a) em instrumentos analgicos; (b) em instrumentos
digitais. A corrente mostrada em S1 e S2 e a diferena entre elas que registrada:
I = Isa - Is1.
Dessa maneira, a corrente capacitiva compensada, pois os seus valores nos dois pontos onde a
corrente amostrada so muito prximos. Os valores das duas correntes esto separados de
dois valores de potenciais por cerca de 50 a 100 mV (que na verdade o valor da amplitude do
pulso superposto).
Na Figura 10b mostra-se a programao de potencial referente a uma instrumentao
digital, onde os pulsos de potencial so superpostos em uma rampa de potencial em forma de
degraus diferentemente de uma rampa de potencial linearmente crescente, como no caso de
instrumentao analgica, mas a amostragem de corrente segue o mesmo esquema da
instrumentao analgica.
A diferena de correntes amostradas em (S1) e em (S2) ser maior ao redor do potencial
de meia onda, onde uma pequena variao de potencial produzir uma grande variao de
corrente. A diferena dessas correntes aumentar at atingir um valor mximo prximo do valor
de potencial correspondente ao potencial de meia onda(E1/2) da polarografia clssica. Aps este
ponto a diferena dos valores de corrente diminuir, voltando ao valor da corrente de fundo e o
polarograma ter uma forma de pico conforme pode ser visto na Figura 11.
Na Figura 12a, os picos de pulso diferencial registrados so de aparelhagem analgica e
o pico da Figura 9b de digital. Na Figura 9b, o potencial correspondente ao ponto mximo do
pico o potencial de pico Ep e serve para identificar a espcie eletroativa, como no caso do E1/2
da polarografia DC. O valor da corrente correspondente ao Ep a corrente de pico Ip, que
proporcional concentrao da espcie eletroativa, permitindo a sua determinao quantitativa.
Para medir a altura do pico e determinar Ip traa-se uma reta tangenciando a base do pico
(Figura 12b).
A seguir mede-se a altura a partir do ponto mximo do pico at a reta tangente base,
perpendicularmente ao eixo x e paralelamente ao eixo y. Essa altura, medida em unidades de
corrente Ip, conforme est mostrado na Figura 12b.
22
FIGURA #%: Polarogramas de pulso diferencial. (a): comparao entre polarogramas DC e de
pulso diferencial para espcies a uma mesma concentrao. As concentraes usadas esto
prximas ao limite de deteco para a polarografia DC. (b): Polarogramas de pulso diferencial
registrado em aparelho digital.
Em razo da corrente capacitiva ser compensada ao registrar-se as diferenas de
corrente, a sensibilidade da tcnica de pulso diferencial (dp) ultrapassa significativamente da
polarografia clssica ou DC, atingindo-se limites de deteco da ordem de 10
-7
a 10
-8
mol/L. No
polarograma da Figura 11a, as duas tcnicas podem ser visualizadas em termos da
sensibilidade. As duas espcies eletroativas apresentam polarogramas DC a concentraes
prximas ao limite de deteco e a tcnica de pulso diferencial mostra os polarogramas nessas
mesmas condies. Pode ser claramente visto na figura, as vantagens da polarografia de pulso
diferencial frente DC, quanto sensibilidade.
23
A resoluo da tcnica tambm melhorada. Na polarografia DC para determinar-se
duas substncias eletroativas simultaneamente elas precisam apresentar uma diferena de
potenciais de meia onda de pelo menos 200 mV. J na polarografia de pulso diferencial uma
diferena entre os potenciais de pico (Ep) de apenas 50 a 100 mV poder ser suficiente para a
determinao simultnea.
A ESCOL3A DE PAR?METROS NA POLAROGRAFIA DE PULSO DIFERENCIAL
Antes de realizar-se uma anlise a escolha de determinados parmetros precisa ser feita.
Um deles, na tcnica de pulso diferencial, o valor da amplitude do pulso a ser usado.
Geralmente escolhe-se um valor entre 10 e 100 mV. Valores tpicos so de 25 mV para sistemas
com um eltron e 50 mV para sistemas com dois eltrons. A amplitude de pulso afeta a corrente
de pico, conforme pode ser visto na equao desenvolvida por Parry e Osteryoung, para um
processo termodinamicamente totalmente reversvel e controlado por difuso:
onde I@ a corrente de pico, .E a amplitude de pulso, A a rea do eletrodo, C a
concentrao da espcie eletroativa, F o faraday, D o coeficiente de difuso, n o nmero de
eltrons, o nmero pi, 3,1416..., t, o tempo de durao do pulso, R a constante dos gases e
T a temperatura absoluta.
Desta equao pode ser claramente visto que Ip proporcional concentrao da
espcie eletroativa e tambm amplitude do pulso. Aumentando-se a amplitude do pulso
aumenta-se o valor da corrente, sendo ento maior a sensibilidade. Entretanto, um aumento da
amplitude provoca um aumento na largura do pico, reduzindo a resoluo. Dois picos adjacentes
no podem ser resolvidos ao menos que a amplitude do pulso seja bem menor do que a
separao entre os dois potenciais de pico.
A escolha da amplitude deve ser um compromisso entre o aumento da sensibilidade e a
perda de resoluo. Da usar-se, de um modo geral os valores tpicos mencionados de 25 mV
para sistemas com um eltron e 50 mV para sistemas com dois eltrons, como j mencionado
anteriormente. Para cada valor de amplitude de pulso ser registrado um polarograma. O que
apresentar um pico melhor definido e com maior valor de Ip, indicar a amplitude a ser
escolhida. Quando a determinao de mais de uma espcie estiver envolvida, ser considerada a
amplitude que fornecer um compromisso entre a maior corrente de pico obtida (que permite
maior sensibilidade) e a melhor separao entre eles (o que permite uma resoluo maior).
Outro parmetro importante a ser escolhido a velocidade de varredura. No caso do
pulso diferencial o valor mximo que pode ser usado de 10 mV s-1. O valor tpico de 5 mV s
-
1
. Se forem usadas velocidades de varreduras maiores que 10 mV s-1 as etapas de potencial
sero muito longas e elas at mesmo podero "pular" o pico, dependendo dessa velocidade.
Nesses casos, haveria, portanto uma perda de resoluo, de modo que ou no haver registros
de corrente em pontos importantes dos picos voltamtricos/polarogrficos (o que afetar as
suas formas e definies) ou, at mesmo, esses picos podero no ser registrados.
Os picos polarogrficos nesta tcnica fornecem uma informao qualitativa, que o valor
do potencial de pico, Ep, e uma informao quantitativa, que a corrente de pico, Ip. Para
construir-se uma curva de calibrao normalmente colocam-se os valores das correntes de pico
contra as concentraes da espcie eletroativa correspondentes a eles. Mas pode-se tambm
usar a rea sob o pico versus a concentrao. Este caso d melhores resultados quando ocorrem
fenmenos de adsoro, por exemplo, alterando a forma do pico de uma medida para outra. A
altura muda, mas a rea permanece constante. No h uma regra definida para a escolha, de
modo que ambas podem ser testadas para verificar-se qual delas a mais adequada, embora no
24
caso de picos com certa sobreposio a medida da corrente de pico (Ip) d melhores resultados
(Figura 13).
FIGURA #+: Polarogramas de pulso diferencial com sobreposio. As linhas tracejadas indicam
a maneira correta de "completar o polarograma para ler as correntes de pico
em termos de altura mxima.
A relao entre a corrente de pico (altura ou rea) e a concentrao da espcie
eletroativa obtida pelo uso da equao de Parry e Osteryoung (equao14) simplificada de
maneira anloga equao de Ilkovic. Mantendo-se todos os parmetros constantes da equao
(14), ela se torna:
onde:
Ip= corrente de pico.
K = constante englobando todos os termos da equao (14) mantidos constante durante
as medidas (exceto C, naturalmente).
C = concentrao da espcie eletroativa.
VOLTAMETRIA DE ONDA 'UADRADA
A polarografia/voltametria de pulso diferencial ainda a tcnica mais usada
presentemente para fins analticos, devido s vantagens apresentadas em relao
detectabilidade e resoluo frente as tcnicas de corrente contnua. Mas, ao lado dela, uma
tcnica muito conveniente do ponto de vista analtico que tem sido incorporada em diversos
instrumentos comerciais a voltametria de onda quadrada de varredura rpida. Ainda que a
voltametria de onda quadrada tenha surgido em 1957 devido aos estudos de Baker, a utilizao
da tcnica era limitada pela tecnologia eletrnica.
Com os progressos da tecnologia analgica e digital, ela passou a ser incorporada nos
polargrafo a partir dos anos 80, principalmente, na sua forma de varredura rpida. Nessa
forma ela tambm chamada de voltametria de onda quadrada de Osteryoung, atribuda ao
nome do pesquisador americano que a desenvolveu.
A tcnica pode ser usada para realizar-se experimentos de um modo bem mais rpido do
que com a tcnica de pulso diferencial, com sensibilidade semelhante ou um pouco melhor, pois
aqui tambm ocorrem compensaes da corrente capacitiva. Um experimento tpico que requer
cerca de trs minutos para ser feito pela polarografia de pulso diferencial pode ser feito em
segundos pela voltametria de onda quadrada.
25
A programao de potencial usada na voltametria de onda quadrada pode ser vista na
Figura 14. Uma onda quadrada simtrica (Figura 14b) superposta sobre uma rampa de
potencial em forma de degraus (Figura 14a) de tal forma que o pulso direto da onda quadrada
coincida com o incio do degrau da rampa (Figura 14c). O pulso reverso da onda quadrada por
sua vez coincide com a metade da etapa da rampa em degraus.
Os parmetros tempo e potencial aplicado so mostrados na Figura 15, onde o
tempo de um ciclo de onda quadrada e tambm de um ciclo da rampa em degraus. A freqncia
da onda quadrada em Hz 1/. Es a amplitude do pulso de onda quadrada em mV, onde 2xEsw
a amplitude pico a pico (amplitude do pico direto ao reverso).
FIGURA #.: Seqencial potencial vs! Tempo (E vs! T) usada em voltametria de onda quadrada.
26
FIGURA #1: Parmetros tempo e potencial aplicado em voltametria de onda quadrada, onde:
= tempo de um ciclo de onda quadrada; 1/ = freqncia da onda quadrada em
Hz; Ew =amplitude de pulso da onda quadrada em mV; E = potencial em mV da
etapa de rampa de potencial e 2Esw = amplitude de pico a pico da onda
quadrada em mV. Eetapa o potencial da etapa da rampa de potencial em
degraus. A velocidade de varredura v para um experimento de voltametria de
onda quadrada pode ser calculada pela equao:
Por exemplo, se for usado um potencial de etapa (Eetapa ) de 2 mV e de 0,01 s
(correspondente freqncia de 100 Hz) a velocidade de varredura ser de 200 mV/s, que
consideravelmente maior que a de 1 at 10 mV/s da tcnica de pulso diferencial.
A medida de corrente na voltametria de onda quadrada feita amostrando-se a mesma
duas vezes durante cada ciclo da onda quadrada, uma vez no final do pulso direto e a outra no
final do pulso reverso. A tcnica discrimina a corrente capacitiva em razo de sua diminuio
mais rpida do que a corrente faradaca, e fazendo-se a amostragem da corrente perto do final
da durao do pulso, como no caso da tcnica de pulso diferencial. A diferena da corrente entre
a duas amostragens registrada em funo do potencial da rampa em degraus. De modo
semelhante tcnica de pulso diferencial, a voltametria de onda quadrada produz picos para
processos faradacos, cuja altura proporcional concentrao da espcie eletroativa.
Devido s rpidas velocidades de varredura usadas na onda quadrada, um
voltamograma inteiro registrado em uma nica gota de mercrio. Os primeiros experimentos
em onda quadrada eram limitados pelo eletrodo gotejante de mercrio (EGM). Neste eletrodo o
mercrio flui constantemente mudando a sua rea superficial na medida em que a gota
formada. A mudana da rea superficial produz inclinaes na linha de base da rampa de
potencial e torna a interpretao dos dados experimentais mais complexas. Na voltametria de
onda quadrada moderna usa-se o eletrodo de mercrio no modo esttico (EMGE, eletrodo de
mercrio de gota esttica ou, do ingls, static mercur" droping electrode, SMDE). Neste eletrodo
a gota formada rapidamente de tal modo que ela permanece de tamanho constante durante
27
todo o tempo despendido para a medida experimental, no apresentando os problemas de rea
superficial que ocorrem com o eletrodo gotejante de mercrio.
Quanto s freqncias usadas em voltametria de onda quadrada, elas encontram-se
tipicamente na regio de 1 a 120 Hz. Dentro desta faixa de freqncias, as medidas podem ser
feitas com velocidades cem vezes maiores ou mais que nas tcnicas de pulso.
VOLTAMETRIA DE REDISSOLU)*O
O desenvolvimento das tcnicas polarogrficas/voltamtricas de pulso diferencial e onda
quadrada permitiram chegar a determinaes analticas da ordem de 10
-7
a 10
-8
mol/L.
entretanto, na moderna anlise de traos necessrio chegar-se a limites abaixo de 10
-8
mol/L,
o que no se consegue apenas utilizando-se medidas polarogrficas/voltamtricas usuais. essas
medidas, tambm chamadas de medidas voltamtricas/polarogrficas diretas, so afetadas pela
chamada corrente de fundo, que a corrente residual j discutida quando tratou-se da
polarografia dc. essa corrente, tambm comentada anteriormente ao tratar-se do
desenvolvimento das tcnicas de pulso e de onda quadrada, apresenta os dois componentes, o
faradaco e o capacitivo. O faradaco, devido a impurezas e oxignio dissolvido pode ser
minimizado pelo uso de reagentes puros e desaerando-se a soluo. O capacitivo minimizado
pela forma de amostragem da corrente. A questo aqui que essa amostragem de fato no zera
a corrente capacitiva, apenas a minimiza. Assim, quando a concentrao atinge valores da
ordem de 10
-8
mol/L cai-se em um problema semelhante aquele que havia na polarografia
clssica: a corrente faradica torna-se muito baixa e fica encoberta pela corrente de fundo,
chegando-se ao limite da tcnica.
Uma possibilidade para resolver esse problema a utilizao de processos de pr-
concentrao da substncia a ser determinada, de maneira que a corrente faradaca possa
aumentar, suplantando a de fundo, e chegar-se a limites de deteco inferiores a 10
-7
- 10
-8
mol/L. Na anlise voltamtrica a pr-concentrao apresenta a vantagem de poder ser feita no
prprio eletrodo de trabalho, sendo, sob este aspecto, bastante fcil e conveniente a sua
utilizao.
VOLTAMETRIA DE REDISSOLU)*O AN8DICA
Uma das tcnicas que se utiliza de processos de pr-concentrao a voltametria de
redissoluo andica( VRA) (do ingls, #nodic $tripping %oltammetr", ASV), muito utilizada na
determinao de metais pesados, uma vez que vrios deles podem ser depositados no eletrodo
de mercrio atravs de eletrlise de solues de seus ons.
Nesta tcnica a etapa de pr-concentrao consiste de uma eletrodeposio a potencial
constante e controlado da espcie eletroativa sobre um eletrodo estacionrio. Esta etapa
seguida por uma etapa de repouso e uma de determinao, sendo que esta ltima consiste na
redissoluo de volta soluo da espcie anteriormente eletrodepositada.
Como exemplo pode ser citado a determinao de ons cobre. Inicialmente ajusta-se o
potencial do eletrodo a um valor suficientemente negativo para reduzir os ons cobre a cobre
metlico, o qual eletrodepositado sobre o eletrodo.
A eletrlise feita por um tempo suficiente e sob agitao constante para concentrar o
cobre na superfcie do eletrodo a partir de um volume relativamente grande da soluo sobre
um volume muito menor do eletrodo, devido pequena superfcie necessria ao eletrodo de
trabalho (que na verdade um microeletrodo). A seguir, deixa-se a soluo em repouso por
alguns segundos para o sistema entrar em equilbrio.
Na etapa seguinte procede-se varredura de potencial para valores mais positivos
(andicos), e o cobre redissolvido retornando soluo, devido sua reoxidao.
28
Ao ocorrer a reoxidao do cobre a corrente variar, e como no caso da reduo, haver
a formao de um pico com o valor de Ep andico praticamente igual ao do Ep catdico para um
sistema reversvel.
A corrente de pico obtida (Ip) proporcional concentrao do cobre, sendo um sinal
analtico correspondente a uma concentrao que estaria abaixo do limite de deteco na
medida voltamtrica/polarogrfica direta.
A pr-concentrao faz com que a concentrao, na gota de mercrio, devido ao seu
volume minsculo, seja muito maior que na soluo, obtendo-se assim um sinal analtico bem
maior relativamente concentrao presente na soluo, explicando-se o aumento da
sensibilidade da tcnica.
As trs etapas envolvidas na voltametria de redissoluo andica sero discutidas a
seguir e esto esquematicamente representadas na Figura 15.
FIGURA #1: Na Figura 15
a
: etapas envolvidas na voltametria de redissoluo andica. (a)
programao E vs t mostrando os tempos de deposio (Td); e potenciais de
deposio Ed, para Cd
2+
e Cu
2+
. (b) Tempo de repouso (T1). (c) etapa de
redissoluo. Na Figura 15b; voltamogramas hipotticos obtidos na etapa da
redissoluo para Cd
2+
e Cu
2+
.
ETAPA DE DEPOSI)*O
Esta etapa principia por escolher o eletrodo de trabalho mais adequado. Os mais usados
so os eletrodos de mercrio de gota pendente, de mercrio de gota esttica, de filme de
mercrio e de carbono vtreo. Praticamente no h diferena entre os eletrodos de mercrio de
gota pendente e de gota esttica, apenas na maneira de amostrar a corrente, nas clulas
29
modernas. Os eletrodos de mercrio so mais vantajosos na determinao de metais pesados
pois muito deles formam amlgamas, produzindo sinais mais reprodutveis do que os depsitos
metlicos formados na superfcie de eletrodos slidos. Os eletrodos estacionrios de gota de
mercrio so usados para concentraes acima de 1 ng/mL e os de filme de mercrio para
concentraes abaixo de 1 ng/mL ("ppb).
O eletrodo de filme de mercrio mais sensvel que o de gota, pois ele tem um volume
bem menor. Mas para concentraes acima de 1 ng/mL ele pode formar compostos
intermetlicos, o que produz interferncias nas determinaes.
Uma vez escolhido o eletrodo, a deposio feita eletroliticamente aplicando-se o
potencial de deposio (Ed) (Figura 13a) durante um determinado tempo e com agitao da
soluo. O tempo de deposio (td) escolhido em funo da espcie eletroativa, ficando
geralmente entre 30 s e 3 min. Tempos muito longos devem ser evitados, pois podem produzir
sinais fora da regio de proporcionalidade entre a corrente e a concentrao. A agitao faz com
que o transporte de massa por conveco mantenha a concentrao da espcie eletroativa junto
superfcie do eletrodo igual do resto da soluo, permitindo um depsito maior do metal em
um dado tempo de deposio do que se o processo de transporte de massa fosse difusional.
Essa agitao deve ser feita velocidade constante e controlada com preciso.
Nesta etapa, ocorrer ento a reduo do metal e conseqentemente a sua deposio
sobre a superfcie do eletrodo. Para o caso de eletrodos de mercrio:
para o caso de eletrodos slidos:
ETAPA DE REPOUSO OU TEMPO DE E'UIL(2RIO
Aps completar-se a deposio do metal, a agitao cessa e durante alguns segundos
deixa-se a soluo em repouso, para que a concentrao do metal depositado homogeinize-se,
entrando em equilbrio na superfcie do eletrodo. No eletrodo de gota de mercrio esse equilbrio
atingido aps a concentrao do metal uniformizar-se pela sua difuso na gota. Isto requer
cerca de 15 a 20 segundos. Para o eletrodo de filme de mercrio este tempo de cerca de 5
segundos , devido ao volume bem menor do filme em relao gota. O tempo correspondente a
esta etapa chamado de tempo de repouso (tr) (Figura 13 a).
ETAPA DE REDISSOLU)*O
esta etapa faz-se a varredura de potencial na direo andica onde o voltamograma ser
registrado, obtendo-se o sinal analtico de acordo com a tcnica voltamtrica escolhida (pulso
diferencial ou onda quadrada, por exemplo). O metal ou metais depositados se redissolvero
quando os seus potenciais de pico forem atingidos (Figura 13b):
As tcnicas voltamtricas mais comuns escolhidas nesta etapa so a de corrente
contnua (DC), chamada aqui de voltametria de varredura linear (LSV, do ingls, "&inear
$tripping %oltammetr"), a voltametria de pulso diferencial e a voltametria de onda quadrada. A
voltametria de varredura linear mais rpida do que a de pulso diferencial (pode-se usar
varreduras de at 1 V/s), mas no discrimina a corrente capacitiva, no sendo adequada para
concentraes abaixo de 20 - 30 ng/mL. Abaixo de 20 ng/mL pode-se usar o pulso diferencial,
que uma tcnica mais sensvel por discriminar a corrente capacitiva. Se a voltametria de onda
30
quadrada de varredura rpida for disponvel, ela pode ser usada pois rene as vantagens do
pulso diferencial e da LSV, resultando em varreduras rpidas com alta sensibilidade.
A ASV apesar de ser uma tcnica muito sensvel e conveniente para a anlise de traos,
ela praticamente restrita a metais que apresentam solubilidade no mercrio, sendo aplicvel
cerca de 30 elementos. Ela pode tambm ser aplicada a alguns compostos inicos e a algumas
substncias orgnicas, que formam compostos pouco solveis com o mercrio e ficam
depositados junto superfcie do eletrodo. Nestes ltimos dois casos a voltametria de
redissoluo normalmente usada com varreduras catdicas, ou seja, no modo de voltametria
de redissoluo catdica (CSV, do ingls, "'at(odic $tripping %oltammetr"), uma vez que as
espcies acumuladas no eletrodo sofrero reduo.
Quanto aos metais, somente alguns poucos podem ser determinados pela voltametria de
redissoluo catdica, tais como Mn e Pb. Eles so determinados na forma de seus xidos
depositados na superfcie de eletrodos slidos, como de carbono ou platina.
VOLTAMETRIA ADSORTIVA POR REDISSOLU)*O
As limitaes apresentadas pela ASV e CSV podem ser removidas atravs da tcnica
voltamtrica chamada de voltametria adsortiva por redissoluo. Esta tcnica foi desenvolvida
mais recentemente, tendo-se em vista justamente superar os limites impostos pela ASV e CSV e
ampliar o uso da voltametria em anlise de traos.
Na voltametria adsortiva, a pr-concentrao feita pela adsoro da espcie eletroativa
na superfcie do eletrodo. No caso de metais isto feito atravs de seus ons complexos.
Adiciona-se ento soluo contendo o on metlico um complexante adequado e o complexo
formado (metal-ligante) que ser acumulado junto superfcie do eletrodo. Dessa maneira a
pr-concentrao no depende da solubilidade do metal no mercrio, como no caso da
voltametria de redissoluo convencional, e metais pouco solveis (no mercrio) podero ser
determinados.
Devido a essas caractersticas, a tcnica tambm aplicvel a um nmero ilimitado de
substncias orgnicas, bastando que elas apenas tenham propriedades superfcie-ativa, para
poderem ser adsorvidas na superfcie do eletrodo de trabalho, e que sejam, evidentemente,
eletroativas.
Quanto a detectabilidade, o limite de deteco pode chegar a valores ao redor de 100
vezes menor dos que os observados na voltametria de redissoluo andica. Podem ser usados
eletrodos de mercrio, onde obtm-se limites de deteco de at 10-11 - 10-12 mol/L, ou
eletrodos slidos, principalmente na regio andica, com limites de deteo da ordem de 10-8 a
10-10 mol/L.
As propriedades adsortivas podem ser verificadas por voltametria cclica, de pulso
diferencial, cronocoulometria, etc. As espcies a serem adsorvidas necessitam possuir um
coeficiente de adsoro maior que 10-4 cm.
O esquema de pr-concentrao utilizado na voltametria adsortiva por redissoluo
(AdSV, do ingls, "#dsorptive $tripping %oltammetr") anlogo ao da voltametria de
redissoluo convencional (ASV).
ETAPA DE PR4CONCENTRA)*O
Os parmetros usados nesta etapa so basicamente os mesmos j discutidos para a
ASV, sendo apenas diferentes os processos de acumulao e de redissoluo.
Na Figura 16 est representado esquematicamente o procedimento usado na voltametria
adsortiva por redissoluo, para a determinao de um metal M na presena do ligante L,
formando o complexo a ser adsorvido MLn. Pode-se usar um potencial anlogo ao potencial de
31
deposio da ASV, o potencial de acumulao (Eac), para obter-se uma pr-concentrao mais
seletiva, embora isso no seja necessrio em muitos casos. Aqui tambm existir um tempo de
acumulao (tac), anlogo ao td da ASV, que tambm cronometrado, durante o qual a espcie
de interesse ser adsorvida sobre a superfcie do eletrodo. As reaes envolvidas nesta etapa
so:
FIGURA #7: Representao esquemtica do procedimento usado na voltametria adsortiva por
redissoluo mostrando as etapas apara a acumulao e redissoluo na
determinao de um metal M
a+
em presena de um ligante L.
ETAPA DE REDISSOLU)*O
Como nesta tcnica o processo envolvido na interface eletrodo-soluo a adsoro, no
h a necessidade de um tempo de repouso ou de equilbrio, como no caso da ASV, onde ocorre
eletrodeposio de metais, e difuso dos mesmos no mercrio. Assim, aps terminar a pr-
concentrao, pode-se proceder a redissoluo, fazendo-se a varredura usualmente na direo
catdica, usando-se a tcnica voltamtrica adequada. Aqui tambm obter-se- um pico, como
no caso da ASV, e a reao eletrdica pode ser representada por:
ou seja, como o metal foi adsorvido, ele ser reduzido a partir do ponto onde o potencial atingir
o valor do potencial de decomposio, e o valor de potencial de pico Ep tambm
correspondente ao E1/2 da onda polarogrfica. Assim, a varredura de potencial aqui ser
catdica, e no andica como no caso da ASV, onde o metal pr-concentrado ser reoxidado.
32
Na voltametria adsortiva por redissoluo tambm h a possibilidade do ligante sofrer
reduo ou oxidao no processo de redissoluo do complexo adsorvido, se o ligante usado for
uma espcie eletroativa. Assim, o seu sinal tambm poder ser usado para a determinao do
metal. Para o caso de substncias orgnicas o processo anlogo.
E0PERIMENTOS PARA VERIFICAR PROPRIEDADES ADSORTIVAS
Para que uma substncia possa ser determinada pela voltametria adsortiva por
redissoluo (AdSV) ela precisa apresentar propriedades de adsoro (superfcie ativa) junto
superfcie do eletrodo de trabalho, alm de ser, evidentemente, eletroativa em uma dada regio
de potencial. Isto pode ser feito usando-se as prprias tcnicas de voltametria de pulso
diferencial e de onda quadrada.
Por exemplo, no caso de um metal, adiciona-se um complexante na soluo e o
complexo formado poder adsorver-se ou no sobre a superfcie do eletrodo. Se for feita a
varredura no sentido catdico em vrios tempos de "espera", ou seja, de acumulao, e o pico
do voltamograma de pulso diferencial ou onda quadrada aumentar a cada vez, de modo anlogo
a esse tipo de estudo com a ASV, indicao de que o complexo estar sendo adsorvido.
Aumentando-se o tempo de acumulao, aumenta-se a adsoro e a concentrao do complexo
na superfcie do eletrodo, aumentando-se ento a corrente de pico a cada etapa, enquanto no
houver saturao da superfcie. Se no houver alterao da altura do pico voltamtrico antes de
uma eventual saturao, o on complexo formado no estar sendo adsorvido e no apresentar
coeficiente de adsoro apropriado ao uso desta tcnica.
Outra tcnica voltamtrica muito empregada para este fim a voltametria cclica
repetitiva (Figura 17).
FIGURA #;: Voltametria cclica repetitiva mostrando voltamogramas
de espcie com propriedades superfcie ativa.
Nesta tcnica realiza-se a varredura de potencial direta e inversa em vrios ciclos
sucessivos observando-se os picos catdicos e andicos da espcie eletroativa. Considerando-se
o exemplo anterior, um metal na forma de um on complexo produzir picos catdicos e
andicos que crescero a cada varredura se houver adsoro. O aumento do pico ocorrer at
evidentemente saturar-se a superfcie do eletrodo. O crescimento dos picos ocorrer
aproximadamente no mesmo potencial se o sistema for reversvel. O pico catdico aumentar a
cada ciclo pelo mesmo motivo que o pico aumenta usando-se as tcnicas de pulso diferencial e
onda quadrada para diferentes tempos de acumulao.
33
A cada ciclo, aumenta-se a concentrao do complexo junto superfcie do metal,
aumentando a corrente do pico, na varredura direta. O metal se deposita ento na superfcie do
eletrodo, dando o pico andico na varredura reversa e aumentando a cada ciclo devido ao
aumento da concentrao do metal reduzido na superfcie do eletrodo.
O TRATAMENTO DE DADOS EM POLAROGRAFIA E VOLTAMETRIA
O tratamento de dados usuais em polarografia e voltametria para fins de anlise
quantitativa, consiste em medir-se a corrente de difuso (no caso da polarografia DC) ou as
correntes de pico no caso de outras tcnicas polarogrficas/voltamtricas como pulso diferencial
e onda quadrada. As correntes obtidas so ento relacionadas s concentraes de solues
padres da espcie eletroativa e concentrao dessa espcie na amostra de interesse. As trs
maneiras mais comuns de se fazer isso sero discutidas a seguir.
MTODO DA CURVA PADR*O
Nesse mtodo, tambm chamado de curva de calibrao ou ainda curva analtica, mede-
se a corrente polarogrfica/voltamtrica de solues padro de vrias concentraes da
substncia em estudo (analito), colocando-se os valores de corrente (corrente de pico, Ip, ou
corrente de difuso, Id) versus os valores de concentrao em um grfico de coordenadas
cartesianas. A curva obtida apresenta um comportamento linear na regio de concentrao de
interesse, passando pela origem no caso das tcnicas clssicas. No caso de tcnicas mais
sensveis, devido as correntes de fundo, ela pode no passar pela origem, o que no afeta o uso
do mtodo. A concentrao calculada pela interpolao da corrente medida da amostra na
curva padro. Isto pode ilustrado na Figura 18.
Geralmente a corrente usada na curva, Ip, a corrente correspondente altura mxima
de pico, mas pode-se tambm usar a rea de pico, quando isto for conveniente.
Esse mtodo o mais comum em qumica analtica. Entretanto, quando se trabalha com
matrizes complexas freqentemente ocorre o chamado efeito de matriz, ou seja, a determinao
da concentrao por interpolao da corrente da amostra lida na curva padro leva a resultados
errados. Isto porque a curva padro feita usando-se solues padres em condies de pureza
e conhecimento das substncias presentes, diferente das condies da matriz, onde se
desconhece a presena de diversas espcies, sendo que vrias delas podem causar
interferncias. No caso de acontecer interferncias, pode-se usar o mtodo da adio de padro.
34
FIGURA #<: Curva padro. O grfico representa a corrente de pico vs. Concentrao do Fe(III)
usando-se a polarografia de pulso diferencial em um eletrlito de suporte base
de citrato em pH 5,00.
MTODO DA ADI)*O DE PADR*O
O mtodo da adio de padro usado com o objetivo de minimizar-se o problema de
efeito de matriz. Como uma matriz complexa geralmente tem espcies presentes que afetam as
propriedades da substncia a ser determinada (por exemplo, amostras contendo substncias
que alteraram a fora inica do meio ou que complexam-se com metais, mudando as
caractersticas polarogrficas/voltamtricas desses metais), a idia realizar as medidas
experimentais nas mesmas condies ou mais prximas possveis das condies da matriz.
Nesse procedimento, a amostra adicionada clula polarogrfica/voltamtrica
juntamente com o eletrlito de suporte e a corrente referente espcie de interesse (analito)
registrada. A seguir, adiciona-se sobre a soluo da amostra uma alquota de alguns microlitros
da soluo padro do analito, de tal modo que a variao do volume total seja desprezvel. Por
exemplo, se a soluo da amostra tiver um volume de 5 mL, adiciona-se uma alquota do padro
de 5 L. Aps a adio do padro, l-se a corrente referente soma da concentrao do analito
mais a concentrao adicional da soluo padro do analito adicionada.
Usualmente adicionam-se pelo menos trs alquotas da soluo padro, registrando-se a
corrente aps cada adio. O volume adicionado escolhido de tal forma a no ocorrer diluio
aprecivel da amostra, o que alteraria as suas condies originais, que aqui s so alteradas
praticamente pela variao da concentrao do analito.
Como as medidas com os padres so feitas em condies muito prximas das condies
da amostra, o efeito de matriz significativamente minimizado, ou seja, as interferncias que
afetam as medidas da corrente referente ao analito presente na amostra, afetaro da mesma
maneira as medidas das correntes referentes ao analito mais as alquotas da soluo padro
sucessivamente adicionadas sobre a amostra.
35
Com os valores obtidos das correntes da amostra e dos padres, constri-se uma curva
corrente vs. concentrao como est mostrado na Figura 19. A corrente da amostra (Ip ou Id,
dependendo da tcnica usada), colocada no eixo "y" para o valor 0 (zero) do eixo "x"'. Os
prximos valores da corrente total de amostra mais padres, para cada adio de padro, so
colocados no eixo "y" e as respectivas concentraes no eixo "x". O grfico resultante deve dar
uma reta. Essa reta extrapolada at cortar o eixo da concentrao (eixo "x") no lado negativo
das coordenadas.
FIGURA #=: Curva de adio de padro utilizada em polarografia/voltametria
O mdulo do valor negativo obtido o valor da concentrao do analito da amostra na
soluo da clula polarogrfica/voltamtrica. Sabendo-se o volume total da soluo na clula
polarogrfica e o volume da amostra original adicionado a ela, calcula-se ento a concentrao
do analito na soluo da amostra original.
Embora o mtodo da adio de padro seja muito vantajoso no caso de matrizes que
contenham espcies no identificadas que possam afetar o sinal medido, tais como substncias
que so adsorvidas na superfcie do eletrodo de trabalho, presena de sais que alteraram a fora
inica do meio, presena de ligantes que formam complexos com ons metlicos, o mtodo da
curva padro considerado mais exato. Assim, se as caractersticas da matriz for reprodutvel e
conhecida, o mtodo da curva padro melhor e deve ser preferido.
MTODO DO PADR*O INTERNO OU (ON PILOTO
Em voltametria/polarografia pode-se tambm usar o mtodo do padro interno para
minimizar o efeito de matriz. Nesse mtodo usa-se uma substncia padro diferente da
substncia a ser determinada (analito) que adicionada amostra. Essa substncia (piloto)
deve ter um potencial de meia onda ou de pico diferente do analito, mas no muito distante,
para que no se use uma varredura de potencial muito longa.
A corrente devido onda ou ao pico polarogrfico registrada para ambos em um
mesmo voltamograma. Assim, assume-se que tudo o que afetar o pico (ou onda) do analito
afetar tambm do mesmo modo o pico (ou onda) do piloto. claro que o mtodo pode ser
aplicado tanto para espcies inicas quanto moleculares. O nome on piloto devido ao mtodo
ter sido desenvolvido originalmente para metais. Na Figura 20 pode-se ver esquematicamente
um polarograma/voltamograma de uma soluo contendo picos do analito e do piloto. A
concentrao do analito determinada pela razo entre a corrente de pico do analito e do on
piloto.
36
FIGURA %>: Mtodo do padro interno ou on piloto usado em polarografia/voltametria
mostrando voltamograma de pulso diferencial com picos do on piloto e da
espcie a ser determinada.
A equao polarogrfica/voltamtrica que relaciona a corrente de pico e concentrao do
padro dada pela equao 17, no caso de medir-se corrente de pico:
onde (ip)p a corrente de pico do padro interno ou on piloto, Ip uma constante (anloga
constante k da equao de Ilkovic) e Cp a concentrao do padro interno ou on piloto. Da
mesma maneira, a equao para o analito torna-se:
onde (ip)x a corrente de pico da substncia desconhecida (analito), Ix uma constante
anloga da equao anterior e Cx a concentrao do analito, a ser determinada. A razo
entre a corrente de pico do on piloto e do analito fica:
daqui chega-se concentrao do analito Cx atravs da equao:
ip/ix chamado de relao do padro interno, Rx. Finalmente a equao (20) se torna:
Rx uma constante vlida para um dado meio e um dado capilar, no caso de usar-se eletrodos
de mercrio no modo gotejante ou esttico. Qualquer alterao de algum parmetro dessas
condies um novo valor de R deve ser determinado.
O mtodo do padro interno ou on piloto considerado muito bom para minimizar
efeitos de matrizes, mas difcil encontrar substncias que apresentem o perfil adequado para
serem usadas como tal. Uma espcie para servir como padro interno necessita ter o seu
potencial de meia onda (ou de pico) relativamente prximo ao do analito e no ter a
possibilidade de ser encontrada como um componente da matriz ou como um contaminante dos
reagentes utilizados.
37
CONSIDERA)AES GERAIS SO2RE MTODOS VOLTAMTRICOS EM
AN/LISE DE TRA)OS
O uso de mtodos voltamtricos em anlise de traos envolve cuidados experimentais do
mesmo modo que outros mtodos analticos utilizados nessas anlises. Assim, importante
realizar a limpeza do material utilizado escrupulosamente, evitar contaminaes, perda do
analito durante os processos de tratamento da amostra, etc. Um problema adicional no caso
especfico de mtodos voltamtricos, que pode ocorrer a formao de compostos
intermetlicos quando se usam eletrodos de mercrio na determinao de metais por ASV.
Todos esses fatores mencionados afetam a exatido e a confiabilidade da anlise e devem ser
minimizados.
MINIMIBA)*O DA CONTAMINA)*O
Na anlise de traos e ultra-traos de metais, o problema de contaminao da amostra
permanente. A confiabilidade e a validade dos dados analticos dependem do grau em que a
contaminao pode ser evitada ou reduzida. Isso remete chamada "boa prtica de
laboratrio", isto , trabalhar sempre atento aos mnimos detalhes referentes aos cuidados
experimentais nas diversas operaes realizadas, como pesagens, limpeza de material, uso
adequado de reagentes (manuseio), estocagem de reagentes, estocagem de amostras, etc.
A vidraria deve ser deixada em cido ntrico 6 mol L
-1
durante vrias horas para
descontaminao, uma vez que o vidro adsorve ons em sua estrutura devido ao efeito da troca
inica. Em seguida, antes de serem usadas, as peas devem ser lavadas vrias vezes com
pores de gua destilada e gua desionizada (pelo menos 6 vezes em cada caso).
Os reagentes usados para a preparao de eletrlitos de suporte e padres devem ser da
mais alta pureza possvel. Alguns desses reagentes so fornecidos comercialmente, outros
necessitam de purificao adicional feita no prprio laboratrio de anlise.
A gua a ser utilizada na preparao de solues de padres e amostras deve ser de alta
pureza. Assim, deve-se usar gua destilada de destiladores de vidro tipo pyrex, destiladores de
quartzo, etc, conforme a necessidade de aplicao. Em geral recomenda-se usar gua
desionizada de alta pureza usando-se o sistema de purificao Millipore Milli-Q.
A contaminao devido a impurezas de reagentes, particulados do ar, e trao de metais
de recipientes e paredes de clulas so os componentes primrios do branco. Devido a isso, o
pr-tratamento da amostra deve ser simplificado. Quanto menor o nmero de etapas no
processo de abertura e preparao da amostra, menor ser a contaminao. Sobre as
contaminaes oriundas de paredes de recipientes feitos de vidro, s vezes prefervel utilizar-
se frascos de teflon ou polietileno, que tem coeficientes de adsoro bem menores que o vidro
em relao a espcies inicas. Solues padres de concentrao abaixo de 10
-3
mol L
-1
jamais
devem ser estocadas. Padres diludos devem ser preparados diariamente.
Quanto ao problema da formao de compostos intermetlicos, isto acontece quando se
usam os eletrodos de mercrio para a anlise de traos de metais por ASV. Ao formar-se um
desses compostos, os picos voltamtricos podem sofrer severas alteraes, diminuindo a altura
ou sofrendo deslocamentos. A causa da formao desses compostos a alta de concentrao no
mercrio de metais que so depositados junto com o analito. Vrios compostos intermetlicos
so conhecidos, incluindo, por exemplo as combinaes: Ag-Cd, Ag-Cu, Cu-Cd, Cu-Ni, Ag-Zn,
Cu-Zn, Co-Zn, Cu-Sb, Cu-Sn, Ni-Sb, etc. Uma interferncia muito comum desse tipo entre
zinco e cobre. Quando eles so depositados simultaneamente pode ocorrer a formao de trs
compostos, CuZn, CuZn2 e CuZn3. Esses compostos produzem picos prximos ao potencial de
pico do cobre, resultando em um aumento da corrente de pico do cobre e uma diminuio da
corrente de pico do zinco.
38
A maneira prtica utilizada para minimizar as interferncias devido a formao de
compostos intermetlicos em anlise de traos por voltametria de redissoluo andica (ASV),
envolve o uso de tcnicas mais sensveis como pulso diferencial ou onda quadrada, em lugar de
varredura linear, pois elas exigem um tempo de deposio menor, diminuindo a concentrao
dos metais no eletrodo e reduzindo a formao desses compostos. Outro recurso utilizado
usar-se o eletrodo de mercrio de gota pendente para concentraes mais elevadas e o de filme
de mercrio apenas para concentraes mais baixas. Assim, de um modo geral, para
concentraes de metais acima de 10 g L
-1
pode-se usar o eletrodo de gota de mercrio, e o de
filme de mercrio para concentraes menores que 10 g L
-1
.
VALIDA)*O DE MTODOS VOLTAMTRICOS
A utilizao de um mtodo analtico exige confiabilidade, ou seja, que o resultado
apresentado pelo mesmo seja exato, o que cientificamente significa que o valor obtido para a
concentrao do analito deve ser verdadeiro dentro do erro experimental. Para isso, quando o
mtodo utilizado em uma dada matriz, h a necessidade de se ter certeza a respeito do
resultado da anlise. Isto feito verificando-se a validez ou validade do mtodo. O procedimento
para essa verificao chamado de validao.
A validao feita envolvendo os parmetros analticos usuais (erro, desvio, desvio
padro, etc) e a obteno do resultado da anlise por mais de uma via analtica e a comparao
entre si dos diferentes valores obtidos. De um modo geral, h trs maneiras comuns para
realizar-se a validao: o mtodo (teste) de recuperao, a anlise de amostra certificada e a
determinao analtica por um segundo mtodo.
TESTE DE RECUPERA)*O
Este procedimento feito enriquecendo-se a amostra com uma quantidade conhecida do
padro utilizado. A amostra mais o padro passam por todo o procedimento de anlise que a
amostra sozinha passou previamente no procedimento usual de anlise. O resultado obtido para
a concentrao do analito com a amostra enriquecida subtrado do resultado obtido da amostra
sozinha, sem o enriquecimento. A diferena deve dar o valor da concentrao do padro
adicionado para o enriquecimento da amostra dentro do erro experimental. O resultado
expresso em termos de porcentagem de recuperao do padro adicionado. O resultado ideal
de uma recuperao de 100%, mas evidentemente o valor obtido aceitvel estar ao redor de
100% dentro do erro experimental do mtodo.
AMOSTRA CERTIFICADA
A amostra certificada uma amostra referente a uma dada matriz, onde vrios de seus
constituintes foram analisados por diversos laboratrios e diversos mtodos, coordenados por
uma instituio. A instituio fornece um certificado sobre o resultado das anlises ao
comercializar a amostra. H amostras certificadas para vrias matrizes, como ligas metlicas,
plantas, material biolgico, solos, etc. A validade do mtodo verificada aqui, determinando-se
o analito para qual o mtodo foi desenvolvido, na matriz certificada, e comparando-se o
resultado obtido com o valor certificado. Esse resultado, para ser aceito, deve concordar com o
valor certificado dentro do erro apresentado por este valor (o valor certificado). Evidentemente,
a matriz referente amostra certificada deve ser do mesmo tipo da matriz estudada.
DETERMINA)*O POR UM SEGUNDO MTODO
Essa via de validao feita usando-se um mtodo diferente do mtodo em estudo. O
ideal que sejam os mais diferentes possveis. Por exemplo, no caso de um mtodo
voltamtrico, que um mtodo eletroanaltico, tratando-se de um metal, pode-se usar para a
validao um mtodo com tcnica espectroanaltica, tal como absoro atmica. Os resultados
obtidos por ambas as tcnicas devem concordar dentro do erro experimental.
39
CONSIDERA)AES GERAIS SO2RE A VALIDA)*O
Como j foi dito, necessrio validar o mtodo o desenvolvido, quando aplicado a uma
matriz, para que o mesmo tenha confiabilidade. Embora possam existir aspectos diversos e
mesmos polmicos sobre essa questo, em geral recomendvel que um dos procedimentos
seja o teste de recuperao. Mesmo que nesse procedimento o pesquisador use os seus prprios
padres, justamente esse aspecto, a certeza que ele pode ter da exatido de seus padres,
que um dos motivos para que ele use essa via. Uma outra razo para usar-se o mtodo de
recuperao que um grande nmero de matrizes com que se trabalha no tem amostras com
certificado disponveis comercialmente, e quando tem podem ser muito caras. Finalmente h o
fato de que nem sempre h a possibilidade de usar-se um segundo mtodo, pela falta de
equipamento. J o teste de recuperao uma via sempre disponvel. Mas, uma s via de
validao pode no ser completamente satisfatria. Assim, recomendvel usar-se pelo menos
duas vias, podendo uma delas ser o mtodo de recuperao, e a outra, um segundo mtodo
com tcnica diferente da do mtodo desenvolvido, ou a anlise de uma amostra certificada,
quando esta estiver disponvel.
Naturalmente, h outros pontos que sempre devem ser considerados em anlises
qumicas, tais como a amostragem e a abertura e tratamento da amostra, que so aspectos
bsicos da qumica analtica e que envolvem todas as tcnicas de anlise.
VOLTAMETRIA C(CLICA
A Voltametria Cclica Rpida (VC ou VCR) uma tcnica muito utilizada in vivo,
desenhada para providenciar uma boa resoluo temporal e qumica. Esta tcnica baseia-se na
aplicao de uma varredura de potencial ao eletrodo, cuja direo varia ao longo do tempo.
Como cada varredura demora em mdia cerca de 20 ms, pode-se aplicar repetidas varreduras
por segundo. A maior diferena entre a voltametria cclica e a voltametria tradicional, a
velocidade da medio e o tamanho dos eletrodos usados (cerca de 8 mm de dimetro).
A VC (ou VCR) permite estudar as propriedades eletrofisiolgicas dos neurnios e,
atendendo ao neurotransmissor que tem sido mais estudado, a DA, a VCR proporciona um bom
estudo dos vrios subsistemas dopaminrgicos e as suas respectivas funes nas desordens
motoras e psicolgicas.
Alm da corrente faradaica, existem outras contribuies de corrente, como resultado do
carregamento da dupla camada (corrente capacitiva) e da existncia de grupos funcionais
eletroativos no eletrodo (como o caso dos xidos nos eletrodos de carbono). Os microeletrodos
em fibra de carbono tm uma linha de base (ou rudo) muito estvel que pode facilmente ser
medida a priori e subtrada da corrente total. A estabilidade da corrente no faradica , alis,
um pr-requisito para os sistemas que empregam a VCR.
O varrimento negativo, seguido da aplicao de um potencial positivo, permite que as
espcies electroctivas retomem ao seu estado de oxidao inicial, evitando o efeito txido que
estas espcies tm sobre os tecidos (neurotoxinas) e ao mesmo tempo, previne a formao de
filmes de xidos sobre o eletrodo (passivao).
APLICA)*O DE TCNICAS VOLTAMTRICAS
A habilidade para fazer medies dos nveis de neurotransmissores em tempo real, com
a voltametria in vivo, permite que muitas questes quanto mecanstica do processo de
neurotransmisso sejam respondidas. Estas anlises fornecem informaes nicas relativas
cintica de administrao de drogas; permitem o estudo de variaes qumicas no crtex,
associadas a alteraes fisiolgicas (como a fome ou a sede) bem como, tm sido utilizado no
estudo de doenas neurodegenerativas como a doena de Parkinson ou Alzheimer.
40
A voltametria uma tcnica que pode ser utilizada para estudar vias bioqumicas e
mecanismos de ao celulares, j que se conseguem produzir eletrodos miniaturizados que
podem ser colocados no fluido extra-celular, com pouca danificao dos tecidos circundantes.
Atualmente tem sido muito utilizada para estudar os mecanismos que esto na base de
regulao dos nveis de neurotransmissores, numa dada regio do crtex, utilizando-se meios
para manipular a secreo e captao desse transmissor. Esta tcnica tem sofrido alteraes ao
longo do tempo, visando ultrapassar alguns problemas, como o caso do bloqueamento do
eletrodo quando este est exposto a constituintes biolgicos e atingir os mais altos nveis de
especificidade, reprodutibilidade e sensibilidade.
Apenas molculas que possam ser facilmente oxidadas ou reduzidas so abrangidas pela
voltametria, como o caso das catecolaminas: dopamina (DA) e noradrenalina (NA) e as aminas
biognicas: serotonina (5-hidroxitriptamina ou 5-HT) e histamina.
Os neurotransmissores so substncias qumicas produzidas pelos neurnios e so
usadas para transmitir informao entre as clulas. Estes so libertados por um neurnio pr-
sinptico para a fenda sinptica, no espao extra-celular e causam uma alterao na membrana
ps-sinptica. Esta clula receptora, pode ser um neurnio com receptores par aos
transmissores, sofrendo uma alterao de potencial, porm, tambm pode ser uma clula
muscular ou uma clula glandular. (Fig.1).
Neurotransmisso a converso de um evento eltrico num evento qumico e
posteriormente noutro evento eltrico. Com o presente trabalho, pretende-se fazer uma
abordagem geral sobre a utilizao da voltametria in vivo para o estudo dos processos da
neurotransmisso.
F!56ra %#: Diagrama simplificado de um terminal nervoso
catecolnico. O neurotransmissor sintetizado e
armazenado em vesculos que fundem com a membrana
libertando o seu contedo para a fenda sinptica. Uma vez
libertos, os transmissores ligam-se aos receptores ps-
sinpticos e aos autorreceptores. A captao e difuso da
catecolamina finda a estimulao da clula ps-sinptica.
O transmissor reciclado e armazenado ou metabolizado.
PERSPECTIVA 3IST8RICA
A voltametria in vivo foi introduzida em 1973
quando Ralph Adams e os seus colaboradores
implantaram, pela primeira vez, um eletrodo miniaturizado
em pasta de carbono, no crtex de um ratinho e
conseguiram detectar material eletroativo. Adams centrou-se mais no estudo das correntes de
oxidao de catecolaminas, porm, deparava-se com o problema de existirem outras espcies
eletroativas, como o cido ascrbico, no mesmo potencial e por estas se encontrarem em
concentraes muito superiores s das catecolaminas.
Gonon desenvolveu outro tipo de eletrodos para detectar as catecolaminas. Nestes caso
ele usou um eletrodo de trabalho em fibra de carbono, associado voltametria de impulso
diferencial, tendo conseguido diminuir o dimetro do eletrodo para 12mm.
Wightman, ao contrrio dos trabalhos anteriores, em vez de procurar detectar os nveis
basais de dopanina no crtex, utilizou a voltametria cclica rpida (VCR) para detectar a
dopamina libertada pelos terminais nervosos, aps estimulao eltrica. A concentrao de
dopamina (e de qualquer outro neurotransmissor) medida afetada pela libertao, captao,
difuso e metabolismo do neurotransmissor.
41
APLICA)*O DE OUTROS TIPOS DE VOLTAMETRIA
Alm da VCR, a cronoamperometria, a amperometria de potencial constante e a
voltametria de pulso diferencial, so tambm tcnicas utilizadas na monitorizao de
neurotransmissores.
Na amperometria de potencial constante, aplicado s um passo de potencial
produzindo-se uma corrente que decai com o tempo. Esta tcnica oferece a melhor resoluo
temporal, porm, no permite distinguir as correntes de oxidao dos vrios analitos,
oferecendo uma fraca seletividade. Esta tcnica tambm muito utilizada na obteno de
senssores e biossensores, visando interesse em neurotransmissores.
Na cronoamperometria aplicado um potencial de onda quadrada, repetitivamente,
medindo-se a variao da corrente com o tempo. O potencial escolhido o suficiente para oxidar
e re-reduzir as espcies de interesse o que tambm evita a acumulao de neurotransmissores
oxidados (como no caso da CVR). Atualmente tem-se utilizado a cronoamperometia ultra-rpida
in vivo, para monitorizar os nveis de 5-HT no meio extra-celular. Para tal introduzem-se os
eletrodos na regio subgranular do gyrus dentado, que uma regio de densa enervao
seretonrgica e, no corpos calosum, o qual est privado do transportador de 5-HT. Neste estudo
utilizou-se meios farmacolgicos, a fuvoxamina que um inibidor da captao de 5-HT, a fim de
ampliar o sinal do transmissor em estudo.
O ELETRODO
Desde o incio do uso da voltametria in vivo at aos dias de hoje, o tipo de eletrodo
utilizado tem sofrido algumas alteraes, nomeadamente no que diz respeito ao seu tamanho,
s tcnicas utilizadas para melhorar a seletividade e a constante de tempo do eletrodo, mas,
tem-se mantido o uso do eletrodo de fibra de carbono. Este pode apresentar a forma de disco,
cilindro ou elipse (Figura 22):
F!56ra %%: Um microeletrodo em fibra de carbono, destinado a medir a oxidao de
nerotransmissores, utilizando uma fibra de carbono (ou um pequeno grupo de fibras)
como elemento condutor.
Os eletrodos tm uma constante de tempo determinada pela propriedade resistiva do
eletrlito e capacitiva da interface do eletrodo com a soluo. Os estudos cinticos esto
limitados por esta constante de tempo uma vez que o eletrodo s consegue medir processos que
durem 1/2 tempo a mais do que a sua constante de tempo. Inicialmente os eletrodos
construdos tinham constantes de tempo da ordem dos 100ms ou mais, atualmente, com os
microeletrodos, atingiram-se os 100 ns, conseguindo-se determinar os tempos de difuso. A
recente introduo dos nanoeletrodos, permite o estudo de mecanismos ultra-rpidos, atingindo-
se constantes de tempo da ordem de 1 ns.
42
O aumento da seletividade e sensibilidade tem sido conseguida com a alterao da
superfcie do eletrodo. Recorre-se por vezes ao uso de eletrocatalizadores ou mediadores, que
podem estar ligados por diversas formas ao eletrodo. O mtodo mais comum na monitorizao
de neurotransmissores, para aumentar a especificidade, a formao de um filme polimrico
com Nafion na superfcie do eletrodo. Dada a sua natureza aninica, consegue-se uma pr-
concentrao de espcies catinicas, como o caso da DA ou NA e repulso de espcies
aninicas, como o ascorbato.
Nas anlises in vivo, deve-se ter em considerao que nas diferentes regies do crtex,
o contedo neuroqumico varia consideravelmente, assim, algum grau de especificidade
conseguido selecionando-se um local alvo apropriado.
Por exemplo, colocando um eletrodo no "striatum" pode-se considerar que a corrente do
oxidao obtida, devida grande concentrao de DA e no de NA, que nesta zona est
presente em baixa quantidade.
Por outro lado, pode-se manipular o contedo do meio extra-celular com uso de
compostos farmacolgicos: pode-se inibir seletivamente a sntese de um dado transmissor ou
bloquear a secreo, diminuindo a sua concentrao no meio extra-celular, ou, pode-se bloquear
a captao do transmissor, produzindo-se um efeito oposto. Isto permite a deteco da DA sem
interferncia do seu principal metabolito. Estas manipulaes farmacolgicas podem causar
alteraes fisiolgicas uma vez que interferem com mecanismos de feedback inerentes ao
processo de neurotransmisso.
Outra forma de melhorar a seletividade in vivo, estimular apenas os axnios dos
neurnios de interesse. Esta aproximao limitada pelo fato do grupo de neurnios que foi
estimulado, poder no representar o que sucede fisiologicamente.
UMA ALTERNATIVA: O ELETRODO DE S(LICA
Os eletrodos de carbono so selados por vidro. Recentemente foram criados novos
eletrodos que procuram responder a alguns problemas, nomeadamente a fragilidade do vidro;
procuram simplicidade de montagem do material no laboratrio e que consigam penetrar mais
fundo no crtex dos animais em estudo. Estes eletrodos so manufaturados com carbono e slica
fundida. Tm boas propriedades mecnicas; so muito fceis de montar num laboratrio, j que
no requerem material especial; exibem uma sensibilidade DA e NA semelhante dos
eletrodos de carbono e podem ser produzidos longos o suficiente para atingir zonas mais
profundas do crtex.
PRO2LEMA DA AN/LISE IN VIVO
A principal diferena entre as anlises eletroqumicas in vivo, em relao anlise de
clulas ou tecidos (brian slice), a distncia entre o local de secreo e o eletrodo. O tamanho
da fenda sinptica muito reduzida, por exemplo, a sinapse dopamnica do striatum, tem uma
fenda de apenas 15 nm, de forma que no se consegue colocar o eletrodo diretamente sobre o
local de secreo. Para se obter correntes de oxidao, necessrio que o neurotransmissor
abandone a sinapse e percorra um caminho at ao eletrodo, por difuso, que dificultado pelas
estruturas celulares.
As correntes medidas refletem ento, as alteraes ocorridas no meio extra-sinptico. A
interaco dos transmissores com receptores e transportadores, diminui a sua concentrao
extra-celular, da que a concentrao seja uma medida da "eficcia" de um neurotransmissor no
crtex.
43
CONCLUS*O RESUMIDA
A voltametria in vivo tem sido utilizada desde que Ralph Adams introduziu pela primeira
vez um eletrodo de trabalho no crtex de um animal. Desde ento esta tcnica tem sofrido as
naturais evolues que decorrem ao longo do tempo. Tem-se mantido o uso do eletrodo em fibra
de carbono, com ligeiras alteraes no sentido de melhorar a seletividade e sensibilidade.
Os mecanismos de neurotransmisso tm sido elucidados principalmente com a VCR.
Ainda recentemente foi desvendado o processo de regulao da captao e secreo da DA ,
com o uso desta tcnica, porm, convm dizer que a voltametria frequentemente confirmada e
complementada por outras tcnicas. O conhecimento bsico do funcionamento anatmico do
sistema nervoso, fundamental para a correta interpretao dos resultados. A microdilise bem
como a cromatografia lquida de alta presso so frequentemente utilizadas em conjunto com a
voltamentria, alargando o espectro de compostos analisados e melhorando a seletividade.
A voltametria tem sido uma ferramenta fundamental no que toca ao esclarecimento de
processos envolvidos na neurotransmisso, mas ainda h muitas barreiras a transpor. Num
futuro talvez possamos contar com nanoeletrodos que consigam atingir os espaos mais
recndidos do nosso crebro, como a fenda sinptica.
2I2LIOGRAFIA UTILIBADA
1. Texto descrito por:
Luiz Manoel Aleixo
Universidade Estadual de Campinas, Instituto de Qumica, CP 6154
13084-862 Campinas, So Paulo - Brasil
OBS: foram feitas alteraes no seu contedo.
2. BRETT, A. M. O., BRETT, C. M. A., 2!oele"troC6D!"a @r!n"@!osE Dtodos e a@l!"aFGes,
Coimbra: Livraria Almedina, 1996.
3. AWS, L.C. TONEY, G.M., DAVIS, D.J.; GERHARDT, G.A. FRAZER, A. In H!Ho
"IronoaD@eroDetr!" Deas6reDents oJ tIe "learan"e oJ eKo5eno6slL a@@l!ed seroton!n
!n tIe rat dentate 5Lr6s, Journal of Neuroscience Methods, 78:1-2:139-150, 1997.
4. FINLAY, Janet M.; SMITH, Gwenn S. A Cr!t!"al AnalLs!s oJ Ne6ro"IeD!"al MetIods Jor
Mon!tor!n5 TransD!tter DLnaD!"s !n tIe 2ra!nME 2000.
5. NATHAN, S. L. et al., AdHan"es !n tIe VoltaDDetr!" AnalLs!s oJ SDall 2!olo5!"allL
ReleHant CoD@o6nds, Analytical Biochemistry 303, 1-16, 2002.
6. SWIERGIEL, Artur H.; PALAMARCHOUK1, Vitaliy S.; DUNN, Adrian J. A NeN Des!5n oJ
CarOon F!Oer M!"roele"trode Jor !n H!Ho HoltaDDetrL 6s!n5 J6sed s!l!"a, Journal of
Neuroscience Methods, in press, 1997.
7. STAMFORD, Jonathan A.; JUSTICE, JR, Joseph B. AnalLt!"al CIeD!strL (68) 359A-366A,
1996.
8. WIGHTMAN, R. M.; MICHAEL, D. J.; Ele"tro"IeD!"al Don!tor!n5 oJ O!o5en!" aD!ne
ne6rotransD!ss!on !n real t!DeE Journal of Pharmaceutical and Biomedical Analysis 19, 33-46,
1998.
9. WU, Qun, et al, Con"6rrent A6tore"e@tor4Med!ated Control oJ Do@aD!ne Release and
U@taPe d6r!n5 Ne6rotransD!ss!on: An In V!Ho VoltaDDetr!" St6dLE TIe ,o6rnal oJ
Ne6ros"!en"eE 22(14):6272-6281, 2002).
10. Neuroscience Clinic http://www.pallidotomy.com/neurotransmitters.html
44
11. LIPOWSKI, Jacek FCIC, LQA"t6al!t "I!D!C6e "anad!enne, janvier, 2000.
In-Vivo Detection Of Neurotransmitters With Fast Cyclic Voltammetry, Mpb Electrodes
Neuroscience Section Medical Building, London
12. (http://www.qmw.ac.uk/~physiol/aboutFCV.html)
13. SAMMUT, Stephen, MUSCAT, RICHARD; TIe 2as!" Pr!n"!@les and A@@l!"at!on oJ Fast
CL"l!" VoltaDDetrL !n tIe Mon!tor!n5 oJ SLna@t!" ne6rotransD!tter oHerJloN and
Ne6ronal Electrophysiological Monitoring
14. (http://www.cis.um.edu.mt/~phcy/symp97/sammut.htm)
15. "http://es.wikipedia.org/wiki/M%C3%A9todos_electroanal%C3%ADticos
REFER&NCIAS INDICADAS PELO AUTOR DO TE0TO
1. Christian, Gary D., AnalLt!"al CIeD!strL, 1st edition, John Wiley & Sons, 1986, New York,
1986.
2. Kolthoff, I.M. e Lingane, J.J., Polaro5ra@ILE Vol. I and Vol. II, 2nd edition, Interscience
Publishers, New York, 1965.
3. Aleixo, L.M., Sitton, M. e Ribeiro, F.A.L., Est6do @olaro5rRJ!"o soOre a deterD!naFSo de
Fe9III: 6t!l!Tando4se a t"n!"a da @olaro5raJ!a de @6lso d!Jeren"!al, Qumica Nova 4th
editon, 2001.
4. Skoog, D. A. e Leary, J.J., Pr!n"!@les oJ Instr6Dental AnalLs!s, 4th edition, Saunders
College Publishing, Philadelphia, 1992.
5. Skoog, D.A., West, D.M. e Holler, J.F., F6ndaDentals oJ AnalLt!"al CIeD!strL, 6th edition,
Saunders College Publishing, Philadelphia, 1992.
6. Vogel, AnRl!se Inor5Un!"a '6ant!tat!Ha, 4a edition, Editora Guanabara Dois S.A., Rio de
Janeiro, 1981.
7. Bond, A.M., Modern Polarographyc Methods in Analytical Chemistry, Marcell Dekker,
Inc., New York, 1980.
8. Bond, A. M., %>> Lears oJ @ra"t!"al ele"troanalLt!"al "IeD!strL: @astE @resent and
J6t6re d!re"t!ons !l6strated OL reJeren"e to tIe on4l!neE on streaD and oJJ4l!ne
deterD!nat!on oJ tra"e Detals !n T!n" @lant ele"trolLte OL HoltaDDetr!" and
@oten"!oDetr!" te"In!C6esE Anal. Chim. Acta 400, 333-379, 1999.
9. Riley, T. e Watson, A., Polaro5ra@IL" and otIer VoltaDDetr!" MetIods, John Willey &
Sons, London, 1987.
10. Wang, J., Str!@@!n5 AnalLs!s 4 Pr!n"!@lesE Instr6Dentat!on and A@@l!"at!ons, VCH
Publishers, Deerfield Beach, 1985.
11. Ivaska, A., Lewenstam, A. e Sara, R.E ConteD@orarL Ele"troanalLt!"al CIeD!strLE
Plenun Press, New York, 1988.
12. Greef, R., Peat, R., Peter, L.M., Fletcher, D. e Robinson, J., Instr6Dental MetIods !n
Ele"tro"IeD!strLE Ellis Horwood Limited, Chichester, 1985.
13. Plambeck, James A., Ele"troanalLt!"al CIeD!strL: 2as!" Pr!n"!@les and A@@l!"at!onsE
John Willey & Sons, New York, 1982.
14. http://www.uc.pt/rnam/artigos/005.htm
45
E0ERC(CIOS DE VOLTAMETRIA
1) Qual a diferena entre as tcnicas: voltametria e polarografia?
2) Que eletrlito suporte?
3) Defina os termos: correntes: catdica, andica, faradica e difusional.
4) EGM: Em que consiste? b) Quais as vantagens? Desvantagem?
5) Fale sobre as tcnicas voltamtricas/polarogrficas D' D' amostrada de pulsos normal e
diferencial, no que se refere a sensibilidade e variao de corrente. Represente-as
graficamente.
6) O que significa o termo desaerao ou purga?
7) Que so reaes nernstiniana?
8) O que voc entende sobre voltametria cclica?
9) O que acontece com um eletrodo a medida que seu potencial se torna mais negativo? E o
inverso?
10) Como acontece o mximo polarogrfico?
11) Quais os processos de velocidade que governam a corrente?
12) Qual a forma de voltametria de onda quadrada? Qual a sensibilidade desta tcnica?
13) Comente sobre a tcnica VRA no que se refere nas etapas de repouso, deposio e de
redissoluo.
14) Em que consiste o tratamento de dados nas tcnicas: polarografia e voltametria?
15) Mtodo de curva padro:
a) O que medido? b) O que representa a curva obtida?
16) Mtodo de adio padro: a) qual o objetivo de seu uso? b) Como feita a adio da
amostra?
17) Mtodo de padro interno: a) como usado? b) Como determinada a concentrao do
analito?
18) Como feita a validao dos mtodos voltamtricos?
19) Como realizado o teste de recuperao em voltametria?
20) Quando dizemos que uma amostra certificada?
22a) Quais as fontes de corrente residual em polarografia de varredura linear? b) Por que as
correntes residuais so menores em polarografia por amostragem de corrente (DC
amostrada)?
Ra) A primeira a corrente de reduo de impurezas presentes na soluo, como ons de metais
pesados presentes na gua destilada, pequenas quantidades de oxignio dissolvido e
impurezas na substncia utilizada como eletrlito suporte.
A segunda denominada de corrente de carregamento, corrente capacitiva ou corrente de
condensador que carrega as gotas de mercrio com relao soluo, podendo ser positiva
ou negativa.
OBS: na varredura linear todas as oscilaes de corrente observadas pelo crescimento e
diminuio da gota de mercrio do uma forma serrilhada toda a variao de corrente.
Rb) Com relao a polarografia DC amostrada, observa-se que as correntes residuais so
menores, isto explicado pelo fato da corrente no ser monitorada continuamente e, sim,
amostrada, ou seja, a corrente s informada ao registrador apenas segundos antes da
gota cair. Desta forma, uma curva em forma de linha e no serra, como na varredura linear
(DC), apresentada.
46
E0EMPLOS ENVOLVENDO VOLTAMETRIA
1) ESTUDO CICLO VOLTAMTRICO DE NITROPRUSSIATO DE SDIO
1
: J.C.SILVA
2
,
L.P.R.PROFETI
3
, N.R.STRADIOTTO
4
, J.F.ANDRADE (orientador)
5
: Departamento de Qumica-
FFCLRP/USP
O nitroprussiato de metais (MFe(CN)5NO) tem sido pouco pesquisado apesar de sua ao
hipotensiva, reduzindo significamente a presso sangunea de pacientes hipertensos e de sua
alta reatividade, podendo ser utilizado como reagente analtico. O nitroprussiato de sdio
usado frequentemente em anlises farmacuticas qualitativamente e quantitativamente. Este
projeto de pesquisa tem como objetivo principal obter os parmetros voltamtricos do
nitroprussiato de sdio para possveis aplicaes analticas. Para obteno desses parmetros,
foram feitos voltamogramas cclicos de vrias concentraes diferentes de nitroprussiato de
sdio ( 2, 4, 6, 8, 10mmol.L
-1
), mantendo a velocidade de varredura constante (100mV.s
-1
).
Tambm foram obtidos voltamogramas cclicos, mantendo a concentrao de nitroprussiato em
4mM e variando a velocidade de varredura ( 20, 50, 100, 200, 500mV.s
-1
). Foram utilizados
(NH4)2SO4, Na2SO4 , K2SO4 a 0.10mol.L
-1
como eletrlito de suporte e um potencial variando ( 0
-0.6V ), para obteno desses parmetros voltamtricos. Como o nitroprussiato anlogo ao
ferrocianeto de potssio, sendo este um sistema conhecido pela literatura e tambm pesquisado
neste laboratrio, por outros colegas, foram efetuados testes para o ferrocianeto de potssio.
Este ser colocado em anexo para comparao com o nitroprussiato de sdio.
1
Projeto
financiado pela CNPq;
2
Bolsista PIBIC/CNPq.
2) COMPORTAMENTO REDOX DE FLAVONIDES
1
: A.A.deLima
2
, W. F. DE Giovani
3
(orientador) -
Departamento de Qumica - FFCLRP/USP
O trabalho consiste no estudo do comportamento eletroqumico e espectroscpico dos complexos
formados entre flavonides como a 3-hidroxiflavona, quercetina, crisina e rutina e ons Cu
2+
,
usando como solventes etanol e metanol.
Em sistemas que apresentam reao qumica acoplada anterior ao processo redox, as
caractersticas dos ciclovoltamogramas so:
a. Com o aumento da velocidade de varredura, o potencial varia para o potencial negativo.
b. O grfico da corrente versus n
1/2
(velocidade de varredura), uma curva exponencial
decrescente.
c. No existe corrente catdica.
Os tens a e b no so observados em nossos ciclovoltamogramas, pois quando se aumenta a
velocidade o potencial aumenta para o potencial positivo e o grfico corrente versus n
1/2
no se
comporta como uma curva exponencial decrescente.
Os valores dos potenciais de oxidao de uma soluo de 3-hidroxiflavona e sulfato de cobre em
meio de metanol, so 0.83, 0.85, 0,87 e 0,90 em 20, 50, 100 e 200 mVs
-1
respectivamente.
Em espectros UV-Visvel dos flavonides; temos os seguintes valores de bandas de absoro
para a rutinaa 249nm, 319nm e 364nm. No complexo com cobre a banda em 364 nm desapare
e aparece uma banda em427 nm; as outras bandas so observadas em 278nm e 321 nm.
Projeto financiado pela PIBIC/CNPQ;
2
Bolsista PIBIC/CNPQ.
47
E0PERIMENTO >#
Determinao dos Valores de Potencial e Nmero de Eltrons do Par
Fe
III
(Cn)6
3-
/Fe
II
(Cn)6
4-
por Voltametria Cclica
Introd6FSo
A voltametria uma tcnica eletroqumica fundamentada na interpretao das relaes
tenso-corrente durante a eletrlise, em meio quiescente, de uma espcie eletroativa muito
diluda, na presena de uma elevada concentrao de um eletrlito inerte. O potencial variado
de modo sistemtico e a curva corrente-potencial obtida chamada de voltamograma!
A aplicao mais comum da voltametria na anlise de qualquer espcie que seja
eletroativa, ou seja, que possa ser oxidada e reduzida. A medida que o eletrodo torna-se mais
negativo, o eletrodo aumenta a sua capacidade de reduzir espcies eletroativas. Inversamente,
quando o potencial torna-se mais positivo, a sua capacidade de oxidar espcies eletroativas
aumenta.
A corrente a medida do fluxo de eltrons devida a transferncia (e
-
) que se processa
quando uma oxidao (perda de eltrons) ou reduo (ganho de eltrons) ocorre sobre uma
superfcie de um eletrodo.
O potencial de reduo formal (E0) de um par reversvel centrado entre o potencial do
pico andico e o catdico, ou seja:
E0 = (Epa + Epc)/2
O nmero (n) de eltrons transferidos na reao de eletrodo para um par reversvel pode
ser determinado por meio da separao entre os potenciais de picos e aproximadamente 0,059
V.
Para um sistema reversvel, a corrente de pico descrita pela equao de Randles-
Sevcik (varredura direta do primeiro ciclo):
Ip = 2,69x10
5
n
3/2
A D
1/2
c v
onde:
Ip =corrente de pico (A).
n = nmero de eltrons
A = rea do eletrodo (cm)
D = coeficiente de difuso (cm
2
/s)
c = concentrao (mol/cm
3
)
v = velocidade de varredura (v/s).
Os valores de Ipa/Ipc 1.
A clula voltamtrica normalmente usada em experimentos eletroqumicos contm trs
eletrodos imersos na soluo a ser analisada:
Eletrodo de Trabalho: onde a reao de interesse ocorre. Ex. (ouro, platina, grafite piroltico,
carbono vtreo);
Eletrodo de Referncia: fornece um potencial estvel, com o qual o potencial do eletrodo de
trabalho comparado. Ex. Eletrodo de Calomelano Saturado (ECS), Prata/ Cloreto de Prata
(Ag/AgCl);
Eletrodo Auxiliar ou Contra Eletrodo: consiste de um material condutor e quimicamente inerte.
Ex. Platina (Pt), Grafite.
48
A soluo a ser analisada contm a amostra dissolvida e um eletrlito de suporte,
numa concentrao de 1,0x10
-3
mol L
1-
, para assegurar uma boa condutividade requerida pela
tcnica e de modo a no interferir nas reaes eletroqumicas dos materiais eletroativos de
interesse. Alguns eletrlitos de suporte so descritos abaixo:
/"!dos: HCl, H2SO4, H3PO4, etc.
2ases: NaOH, KOH, NH4OH, etc.
TaD@Ges: citrato, fosfato, borato, etc.
As tcnicas voltamtricas so aplicadas na determinao de elementos eletroativos a
nvel de traos, apresentando resultados com boas preciso e exatido.
Este experimento objetiva utilizar a tcnica de voltametria cclica (VC) para determinar
os valores de potencial (Ep) e nmero de eltrons (n) do par Fe
III
(CN)6
3-
/Fe
II
(CN)6
4-
. Pretende-se
ainda avaliar os efeitos da velocidade de varredura, concentrao das espcies eletroativas e
reversibilidade.
Pro"ed!Dento
Preparar uma soluo estoque de K3Fe(CN)6 1,0 mM em K2SO4, 0,1 mol/L.
Efetuar um pr-tratamento da superfcie do eletrodo de trabalho atravs de um
polimento com alumina, lavagem com gua e ultra-som.
Preencher a clula eletroqumica com K3Fe(CN)6 at imerso dos eletrodos.
Desaerar a soluo com gs inerte (N2 ou argnio) por aproximadamente 10 min.
Durante a desoxigenao, ajustar os parmetros do equipamento (com eletrodo de
trabalho desconectado): Ei = -100 mV; Ef = +600 mV; direo = negativa para a
positiva; v = 100 mV/s.
Conectar o eletrodo de trabalho.
Iniciar a varredura para obter o voltamograma cclico (VC) do par redox
Fe
III
(CN)6
3-
/Fe
II
(CN)6
4-
.
Os efeitos de "v so observados, repetindo um experimento VC para cada velocidade
(25, 50, 75, 100mV/s).
TrataDento de dados
Do voltamograma cclico obtido para o par Fe
III
(CN)6
3-
/ Fe
II
(CN)6
4-
, em eletrodo de
platina, determinar os valores de Ep (potencial de pico do par Fe
+2
/Fe
+3
).
2!Ol!o5raJ!a Cons6ltada
Kissinger, P. T. e Heineman, W. R. LaOoratorL Te"In!C6es !n Ele"troanalLt!"al CIeD!strL ,
Dekker, New York. 1984.
Sawyer, D. T.; Heineman, W. R.; Beebe, J. M. CIeD!strL EK@er!Dents Jor Instr6Dental
MetIods, John Wiley & Sons, New York, 1984.
Skoog, D. A.; Holler, F. J. e Nieman, T. A. Pr!n"@!o de AnRl!se Instr6Dental, Artmed
Editora S.A, So Paulo, 2002.
49
E0PERIMENTO >%
DeterD!naFSo do Poten"!al de OK!daFSo da Do@aD!na
@or VoltaDetr!a de Onda '6adrada
Introd6FSo
A Voltametria de Onda Quadrada (SWV) a mais sensvel das tcnicas tradicionais de
pulso. Nesta tcnica a medida de corrente realizada com velocidades de varredura maiores que
100mV/s, e as medidas de corrente sendo realizadas apenas ao final do pulso de potencial, onde
a magnitude da corrente capacitiva j est minimizada. Como vantagens pode ser citada a
formao de ondas quadradas sobrepostas de forma a construir degraus de potenciais e o fato
de que a varredura completa de potencial, no intervalo de trabalho, pode ser feita mais
rapidamente.
A tcnica consiste em uma amostragem da corrente no final de cada um dos pulsos. Em
sistemas reversveis as correntes reversas so significantes, assim as correntes de sada so
maiores do que as correntes diretas ou reversas. Essa uma das razes para que a SWV
apresente maior sensibilidade do que as tcnicas: voltametria de pulso diferencial e polarografia.
A SWV apresenta, em relao voltametria cclica, uma maior capacidade de deteco de
pequenas quantidades de compostos, pois no gera corrente capacitiva constante durante a
medida. A maior sensibilidade e velocidade dessa tcnica tm aumentado seu uso em
determinaes quantitativas de espcies eletroativas em soluo.
O experimento proposto objetiva utilizar a tcnica de voltametria de onda quadrada
(SWV) para determinar o potencial de oxidao da dopamina, [DA] = 1x 10
-4
mol/L em soluo
tampo fosfato pH 7.
Pro"ed!Dento
Antes de comear seu experimento seu professor ou monitor lhe mostrar a estrutura da
dopamina e falar sobre sua importncia na pesquisa cientfica, do ponto de vista clnico e
farmacutico.
Os experimentos eletroqumicos sero realizados em uma cela eletroqumica (Figura 1)
de trs compartimentos contendo:
Eletrodo de Trabalho: Pasta de carbono
Eletrodo de Referncia: Prata /Cloreto de Prata (Ag/AgCl) ou Calomelano Saturado (ECS).
Eletrodo Auxiliar ou Contra Eletrodo: Platina (Pt)
50
F!56ra #: D!a5raDa da "l6la eletroC6D!"a
(1) Entrada para o eletrodo de trabalho
(2) Entrada para o eletrodo auxiliar
(3) Entrada para o eletrodo de referncia
(4) Entrada Scalp
(5) Tampas de teflon
(6) Cela
F!56ra %: D!a5raDa do eletrodo de
traOalIo
(1) Fio de Platina
(2) Tubo de Vidro
(3) Suporte do Eletrodo
(4) Resina
(5) Cavidade para o eletrodo modificado
com pasta de carbono
)reparo da pasta de carbono
Misturar 95 mg de p de grafite com 5 mg de Ftalocianina de Ferro (FePc) e ir
adicionando, aos poucos, 100 L de leo mineral (Nujol
), para que a pasta de
carbono adquira uma consistncia. Esta colocada em uma cavidade na extremidade
de um tubo de vidro (4 mm de dimetro interno) com uma profundidade de 1mm, e
pressionada sobre uma folha de papel sulfite at obter uma superfcie lisa.
*so do aparel(o
Colocar 10 mL da soluo na clula eletroqumica e, aps inserir os eletrodos nos locais
indicados (Figura 1) colocar sob fluxo de nitrognio por 3 min, para que a soluo seja
desaerada. Ligar o potenciostato nas seguintes condies: potencial de 0 a 600 mV,
corrente de 10A, 4 mV de degrau de potencial, 25 mV para a amplitude da onda
quadrada e freqncia de 15 Hz empregando assim, a tcnica SWV, usando um
potenciostato BAS CV-50W, acoplado a um microcomputador
TrataDento de dados
Analisar o voltamograma em relao corrente demonstrando o que indica a presena
da dopamina e mostrar o pico de potencial registrado referente oxidao da
dopamina e em relao reduo do aminocromo.
51
2I2LIOGRAFIA CONSULTADA
Wang, J. AnalLt!"al Ele"tro"IeD!strL, John Wiley & Sons, New York, 2000, p.118-127, 171-
202.
Eggins, B. R. Biosensors: An Introd6"t!on, John Wiley & Sons, New York, 1997.
Sotomayor, M.D.P.T.; Kubota, L.T., '6D!"a NoHa, 2002E %1E 123.
Sotomayor, M.D.P.; Tanaka, A.A.; Kubota, L.T.; AnalMCI!DMA"ta, 2002, 455, 215.
Hasebe, Y.; Akiyama, T.; Yagisawa, T.; Uchiyama, S.; TalantaE 1998, 47, 1139.
Osteryoung, J. e ODea, J. Ele"troanalLt!"al CIeD!strL, Ed. A. J. Bard, Dekker, New York, Vol.
14, 1986, pp. 209-308.
Skoog, D. A., Holler, F. J. e Nieman, T. A., Pr!n"@!o de AnRl!se Instr6Dental, Artmed
Editora S.A, So Paulo, 2002.
52
E0PERIMENTO >+
DeterD!naFSo da Corrente de OK!daFSo do Cate"ol
@or CronoaD@eroDetr!a
Introd6FSo
A cronoamperometria uma tcnica eletroqumica de pulso, que consiste em aplicar um
potencial constante e analisar a corrente resultante que pode ser identificada atravs de anlise
de sua variao com o tempo. A curva corrente versus tempo chamada cronoamperograma.
Esta tcnica permite utilizar sensores amperomtricos os quais so promissores para o
desenvolvimento de tcnicas analticas versteis na determinao de compostos fenlicos, como
por exemplo, o catecol.
Este experimento objetiva utilizar a tcnica de cronoamperometria para determinao da
corrente de oxidao do catecol em soluo tampo fosfato.
Pro"ed!Dento
Os experimentos eletroqumicos sero realizados em uma cela eletroqumica (Figura 1)
de trs compartimentos contendo:
Eletrodo de trabalho: Pasta de carbono
Eletrodo de referncia: Prata/Cloreto de Prata (Ag/AgCl) ou Calomelano Saturado (ECS).
Eletrodo auxiliar ou contra eletrodo: Platina(Pt)
F!56ra %: D!a5raDa da "l6la eletroC6D!"a
(1) Entrada para o eletrodo de trabalho
(2) Entrada para o eletrodo auxiliar
(3) Entrada para o eletrodo de referncia
(4) Entrada scalp
(5) Tampas de teflon
(6) Cela
F!56ra +: D!a5raDa do eletrodo de
traOalIo
(1) Fio de platina
(2) Tubo de vidro
(3) Suporte do eletrodo
(4) Resina
(5) Cavidade para o eletrodo modificado
As medidas eletroqumicas sero efetuadas em um potenciostato acoplado a um
microcomputador, empregando a tcnica de cronoamperometria.
53
)reparo da pasta de carbono
Misturar 100 mg de p de grafite com 15 mg de ftalocianina de mangans (MnPc), 90 mg de
histidina (His) e 1,0 mL de soluo tampo fosfato em concentrao 0,1 mol L
-1
e em pH 6.
Adicionar 100 L de leo mineral (Nujol) para que a pasta de carbono possa adquirir
consistncia.
Colocar a pasta de carbono modificada em uma cavidade na extremidade de um tubo de vidro
(4 mm de dimetro interno) com uma profundidade de 1 mm e pressionada sobre uma folha
de papelpara obteno de uma superfcie lisa.
*so do aparel(o
Colocar 10 mL da soluo tampo fosfato pH 7 na cela eletroqumica e, aps inserir
os eletrodos nos locais indicados (Figura 1), ligar o potenciostato nas seguintes
condies: potencial de 0 mV, corrente de 10 A e tempo de 600 s.
Adicionar perxido de hidrognio, para aumentar a sensibilidade do sistema. Convm
lembrar que na superfcie do eletrodo ocorrer a reao de reduo do perxido e
oxidao do MnPc.
Efetuar a varredura de corrente e esperar estabilizar para em seguida ser adicionado
100 L de catecol 1,0 x 10
3-
mol/L. Fazer adies de 50 em 50 s de soluo de catecol,
perfazendo um total de 7 pontos. Ao adicionar o catecol este ser oxidado.
TrataDento de dados
Fazer a curva analtica (variao da corrente vs concentrao) de catecol adicionado,
visando obter o melhor coeficiente de correlao.
2!Ol!o5raJ!a Cons6ltada
Rosatto, S. S.; Kubota, L. T. e Oliveira Neto, G. AnalM CI!DM A"ta, +=>E 65, 1999.
Lima, A.W.O., Nascimento, V.B., Pedrotti, J.J. and Angnes, J.J. AnalM CI!DM A"ta, +1., 325,
1997.
Sotomayor, M. D. P. T.; Tanaka, A. A.; e Kubota, L. T. AnalM CI!DM A"ta, %#1, 223, 2002.
B. Wang and S. Dong, ,MEle"troanalM CIeD. .<;, 45, 2000.
Skoog, D. A.; Holler, F. J. e Nieman, T. A. Pr!n"@!o de AnRl!se Instr6DentalE Artmed
Editora S. A.,So Paulo, 2002.
54
E0PERIMENTO >.
DeterD!naFSo Polaro5rRJ!"a de CI6DOo 9II:
Introd6FSo
A polarografia iniciou com Heyrovsky que usou a propriedade do mercrio dissolver
alguns metais e ao mesmo tempo a de ser um bom condutor de eletricidade. Esta tcnica um
mtodo eletroqumico baseado no registro da variao que ocorre quando a voltagem aplicada
atinge o potencial no qual as espcies de interesse reagem eletroquimicamente. O mtodo
consiste em uma eletrlise onde o eletrodo de trabalho utilizado um micro eletrodo gotejante
de mercrio.
O experimento objetiva realizar um experimento polarogrfico, para determinao de Pb
(II) em soluo.
Pro"ed!Dento
Colocar 5 mL de gua e 0,5 mL do eletrlito suporte, NaNO3 0,1 mol/L, na clula
eletroqumica e desaerar por 8 min com gs nitrognio, sob agitao constante.
Durante o tempo de desaerao, ajustar os parmetros do sistema voltamtrico
polarogrfico, acompanhado de um agitador magntico e um registrador X-Y, nas
seguintes condies:
Sensibilidade = 0,5 mA.
Potencial inicial = 0.0 V.
Potencial final = -1,0 V.
Tempo de gota = 1 segundo.
Registrador X = 100 mV/cm.
Registrador Y = 100 ou 250 mV/cm.
Velocidade de varredura 10 mV/s.
Iniciar a varredura de potencial apertando a tecla "scan. Este experimento mostra o
polarograma apenas do eletrlito suporte.
Em seguida adicionar algumas gotas de Triton-X-100 que um supressor de mximo
polarogrfico. Sua adio evita deformao no polarograma do on Pb (II).
Adicionar uma alquota de 50 L de soluo estoque da soluo de Pb (NO3)2 0,1 mol/L
para a clula eletroqumica. A soluo resultante ter uma concentrao de 1 x 10
-3
mol/L. Realizar ento, nova varredura nas condies j determinadas, usando as
tcnicas polarogrficas de direct corrent +D', e diferencial de pulso.
Adicionar mais quatro alquotas de Pb (NO3)2, de 50 L cada, clula eletroqumica.
Para cada uma realizar as varreduras nos modos anteriores, obtendo-se polarogramas
correspondentes s concentraes de 2 x 10
-3
mol/L, 3 x 10
-3
mol/L, 4 x 10
-3
mol L
-1
e
5 x 10
-3
mol/L de Pb(NO3)2.
TrataDento de dados
55
Os polarogramas devero ser enumerados e serem usados para determinar a corrente
de difuso, o potencial de meia onda e o nmero de eltrons.
Fazer uma tabela contendo duas colunas: concentrao de Pb (NO3)2 em mol/l e
corrente (A), para os polarogramas no modo DC.
Construir o grfico correspondente Equao de Ilkovic e determinar os valores dos
coeficientes angular, linear e de correlao.
Escrever a equao da reta.
O2SERVA)*O: este roteiro de prtica foi gentilmente cedido pela Prof
a
. Dr
a
. Aldala L.
B. Marques.
2!Ol!o5raJ!a Cons6ltada
Ohlweiler, O. A. Qumica Analtica Quantitatia, 2
a
edio. Livros Tcnicos e Cientficos, Rio
de Janeiro. 1976.
Skoog, A. D.; West, D. M., e Holler, F. J. !undamentals o" Analytical Chemistry. 6
a
edio
Saunders College Publishing, New York, 1981.
Christian, G. D. Analytical ChemistryE John Wiley & Sons, 4
a
edio, New York. 1986.
Skoog, D. A., Holler, F. J. e Nieman, T. A. Princpio de An#lise InstrumentalE Artmed
Editora
S.A., 2
a
edio, So Paulo, 2002.
56
Potrebbero piacerti anche
- Eletroanálises: aspectos teóricos e práticosDa EverandEletroanálises: aspectos teóricos e práticosNessuna valutazione finora
- Rel - Eletrodeposição de CobreDocumento14 pagineRel - Eletrodeposição de Cobreastéria_mendonçaNessuna valutazione finora
- Potenciometria: aspectos teóricos e práticosDa EverandPotenciometria: aspectos teóricos e práticosNessuna valutazione finora
- Voltametria: Conceitos e TécnicasDocumento21 pagineVoltametria: Conceitos e TécnicasGabrieli Bernardi100% (1)
- Roteiro Prática VoltametriaDocumento2 pagineRoteiro Prática VoltametriaFarmacia 102Nessuna valutazione finora
- Espectroanalitica - Emissao MolecularDocumento38 pagineEspectroanalitica - Emissao MolecularflavianesantosNessuna valutazione finora
- QAI, COV, Poluição e Nano TiO2: estudo de fotodegradação de benzeno por nano TiO2 em revestimento cerâmico comercial e assistida por radiação ultravioletaDa EverandQAI, COV, Poluição e Nano TiO2: estudo de fotodegradação de benzeno por nano TiO2 em revestimento cerâmico comercial e assistida por radiação ultravioletaNessuna valutazione finora
- Relatório FluorimetriaDocumento13 pagineRelatório FluorimetriaLucas GamaNessuna valutazione finora
- 01-Fluorescência e FosforescênciaDocumento2 pagine01-Fluorescência e Fosforescêncialevi_santosNessuna valutazione finora
- Aula 12 - CoulometriaDocumento17 pagineAula 12 - Coulometriadigo_jp67% (3)
- Exercicios 2018Documento2 pagineExercicios 2018lucas ronieryNessuna valutazione finora
- Relatorio 5 - CompletoDocumento15 pagineRelatorio 5 - CompletoFernanda BonfimNessuna valutazione finora
- Fosforescência e Fluorescência PDFDocumento30 pagineFosforescência e Fluorescência PDFDulce GabrielNessuna valutazione finora
- Enunciados Exemplos - AdsorçãoDocumento30 pagineEnunciados Exemplos - AdsorçãoFelipe Castro100% (1)
- Relatório de Eletroanalitica Eletrodo de VidroDocumento7 pagineRelatório de Eletroanalitica Eletrodo de VidroGabrielle CristineNessuna valutazione finora
- Relatório Prática 4Documento6 pagineRelatório Prática 4Milena LimaNessuna valutazione finora
- Espectroscopia de Luminescência PDFDocumento9 pagineEspectroscopia de Luminescência PDFmilhosoNessuna valutazione finora
- Lista de Exercícios de Ligações, Termodinâmica e Oxidação-Redução (Recuperado)Documento5 pagineLista de Exercícios de Ligações, Termodinâmica e Oxidação-Redução (Recuperado)S.V.W.M ELÉTRICANessuna valutazione finora
- 5.1. Sebenta Haletos de AlquiloDocumento10 pagine5.1. Sebenta Haletos de AlquiloMariana IsabelNessuna valutazione finora
- Experimento 5 Titulação Potenciometrica Ácido BaseDocumento2 pagineExperimento 5 Titulação Potenciometrica Ácido BaseGeilson SilvaNessuna valutazione finora
- PolarimetriaDocumento6 paginePolarimetrialeandrinholdr6365Nessuna valutazione finora
- Métodos Instrumentais de Análise QuímicaDocumento32 pagineMétodos Instrumentais de Análise QuímicaAlexander DomnicNessuna valutazione finora
- LISTA 1 - Tranf Calor - Exerc Revisão INTRODUÇÃODocumento5 pagineLISTA 1 - Tranf Calor - Exerc Revisão INTRODUÇÃOAugusto Caballero BernardoNessuna valutazione finora
- Relatório de Aula Prática - Determinação de Parâmetros Cinéticos de Reações HomogêneasDocumento32 pagineRelatório de Aula Prática - Determinação de Parâmetros Cinéticos de Reações HomogêneasRicardo Engenharia Ribeirão100% (1)
- Lista 3 Fisico-Químia UERJDocumento2 pagineLista 3 Fisico-Químia UERJGalenoNessuna valutazione finora
- Elaboracao Aula 5 - Regra Da Alavanca PDFDocumento50 pagineElaboracao Aula 5 - Regra Da Alavanca PDFFelix Ferreira100% (2)
- Eletroquimica Relatorio 2Documento9 pagineEletroquimica Relatorio 2CMEI MDMCNessuna valutazione finora
- EXP - 7 - Energia de Ativação PDFDocumento6 pagineEXP - 7 - Energia de Ativação PDFMateus Freitas PaivaNessuna valutazione finora
- Experimento 03 - Determinação Da Condutividade de Solução Aquosa de Ácido Acético Com Diferentes ConcentraçõesDocumento11 pagineExperimento 03 - Determinação Da Condutividade de Solução Aquosa de Ácido Acético Com Diferentes ConcentraçõesFelipe Marcassa100% (1)
- Lista 02 - Volumetria de ComplexacaoDocumento2 pagineLista 02 - Volumetria de ComplexacaoAna Raquel MartinsNessuna valutazione finora
- Apostila de Cinética Química - Eadqui047Documento50 pagineApostila de Cinética Química - Eadqui047Leila MurielNessuna valutazione finora
- Viscosidade de Líquidos - RelatórioDocumento7 pagineViscosidade de Líquidos - RelatórioDe Paulo JoãoNessuna valutazione finora
- Roteiro Experimental de PilhaDocumento4 pagineRoteiro Experimental de PilhaFernanda RibeiroNessuna valutazione finora
- Relatório 1 Marli - Espectrofotometria Uv - VisDocumento26 pagineRelatório 1 Marli - Espectrofotometria Uv - VisNathalie Vieira0% (1)
- Aula de VoltametriaDocumento86 pagineAula de Voltametriamikey123454528638790% (1)
- RELATÓRIO CAA - Thaynara CoutinhoDocumento42 pagineRELATÓRIO CAA - Thaynara CoutinhoThaynara CoutinhoNessuna valutazione finora
- 3 Exercicicos 2 Gases Reais PDFDocumento2 pagine3 Exercicicos 2 Gases Reais PDFVanilson SertãoNessuna valutazione finora
- 05 Espectroscopia de Absorção Atômica e MolecularDocumento49 pagine05 Espectroscopia de Absorção Atômica e MolecularDomenique Delmiro100% (1)
- Aula Eletroanalítica 2008Documento33 pagineAula Eletroanalítica 2008fran_andradeNessuna valutazione finora
- Aula Cinética QuímicaDocumento33 pagineAula Cinética QuímicaBixoFelipeGomesNessuna valutazione finora
- Slides Uv-Vis - EQ PDFDocumento49 pagineSlides Uv-Vis - EQ PDFSilvio Marinho100% (1)
- Determinação Da Energia de Ativação de Uma Reação Iônica em Meio AquosoDocumento10 pagineDeterminação Da Energia de Ativação de Uma Reação Iônica em Meio AquosoAlexsander LopesNessuna valutazione finora
- CondutânciaDocumento19 pagineCondutânciaAmanda CunhaNessuna valutazione finora
- QuimFisicaTCap2 (Noções de Termodinâmica de Líquidos e Soluções) (Exercícios)Documento2 pagineQuimFisicaTCap2 (Noções de Termodinâmica de Líquidos e Soluções) (Exercícios)juliobernardiNessuna valutazione finora
- Seminário Espectroscopia Massas 1Documento24 pagineSeminário Espectroscopia Massas 1kauevergaraNessuna valutazione finora
- Efeito Íon ComumDocumento25 pagineEfeito Íon ComumAlessandrafsaNessuna valutazione finora
- Determinação Da Concentração de ProteínasDocumento6 pagineDeterminação Da Concentração de ProteínasElizael Goncalves100% (1)
- Determinacao de FerroDocumento5 pagineDeterminacao de FerroLetícia LimaNessuna valutazione finora
- Cap 14Documento51 pagineCap 14danieleportuNessuna valutazione finora
- Unidade II - Átomos, Moléculas e ÍonsDocumento4 pagineUnidade II - Átomos, Moléculas e ÍonsMatheus EduardoNessuna valutazione finora
- Fotometria de ChamaDocumento30 pagineFotometria de ChamaSandy MaraNessuna valutazione finora
- Aula de EletrogravimetriaDocumento19 pagineAula de EletrogravimetriaSarah Mariana0% (1)
- Relatório 6Documento15 pagineRelatório 6Fernando HamerskiNessuna valutazione finora
- Determinação de Cobre Na CachaçaDocumento3 pagineDeterminação de Cobre Na Cachaçaelainer2pNessuna valutazione finora
- Complexos 6 Teoria Do Campo CristalinoDocumento29 pagineComplexos 6 Teoria Do Campo CristalinoRenan Soares100% (1)
- Experimento 08 - Cinética Química: Velocidade Das Reações QuímicasDocumento10 pagineExperimento 08 - Cinética Química: Velocidade Das Reações QuímicasHudson Silva100% (1)
- PolarimetriaDocumento5 paginePolarimetriaingritirsNessuna valutazione finora
- EXERCICIOs de Voltametria Com Respostas 1Documento4 pagineEXERCICIOs de Voltametria Com Respostas 1WalasJoãoNessuna valutazione finora