Potrebbero piacerti anche
- Metabolismo del sistema digestivo, del hígado, de la vesícula y de las vías biliares: En condiciones de salud y en las enfermedadesDa EverandMetabolismo del sistema digestivo, del hígado, de la vesícula y de las vías biliares: En condiciones de salud y en las enfermedadesNessuna valutazione finora
- Aterogenesis y Diabetes-IR e Hiperinsulinemia CompensatoriaDocumento5 pagineAterogenesis y Diabetes-IR e Hiperinsulinemia CompensatoriaEleana De La HozNessuna valutazione finora
- sct221ckDocumento8 paginesct221ckVerónica UriósteguiNessuna valutazione finora
- Señales Intracelulares Control GlucosaDocumento13 pagineSeñales Intracelulares Control GlucosaKevin San Martin SanchezNessuna valutazione finora
- Adiponectina y Sus Efectos Pleiotrópicos en El Sistema Cardiovascular. RevisiónDocumento8 pagineAdiponectina y Sus Efectos Pleiotrópicos en El Sistema Cardiovascular. Revisión21121c0514Nessuna valutazione finora
- Defining The Underlying Defect in Insulin Action in Type 2Documento24 pagineDefining The Underlying Defect in Insulin Action in Type 2JOEL ANDRE SANCHEZ GILNessuna valutazione finora
- Caso Clínico - Avance 2 - Brupo B5Documento10 pagineCaso Clínico - Avance 2 - Brupo B5JANIN GEORGETTE RAMIREZ CASTRONessuna valutazione finora
- InsulinaDocumento2 pagineInsulinaDalia López CruzNessuna valutazione finora
- Ensayo Diabetes Mellitus 2Documento2 pagineEnsayo Diabetes Mellitus 2Vale MoralesNessuna valutazione finora
- Macrófagos, Inflamación, Tejido Adiposo, Obesidad y Resistencia A La InsulinaDocumento8 pagineMacrófagos, Inflamación, Tejido Adiposo, Obesidad y Resistencia A La Insulinaphouses101100% (1)
- Artículos de Revisión: ResumenDocumento11 pagineArtículos de Revisión: ResumenFernando PinoNessuna valutazione finora
- Seminario BioquimicaDocumento22 pagineSeminario BioquimicaAngi Carolina Valdelamar MonteroNessuna valutazione finora
- Hormonas Pancreáticas y AntidiabéticosDocumento33 pagineHormonas Pancreáticas y AntidiabéticosKidderleng Sotelo FittoriaNessuna valutazione finora
- Vías Inflamatorias en La Resistencia A La InsulinaDocumento4 pagineVías Inflamatorias en La Resistencia A La InsulinaRuth Vargas ChaNessuna valutazione finora
- Mecanismos de SeñalizaciónDocumento11 pagineMecanismos de SeñalizaciónAngie Zarraga PinedaNessuna valutazione finora
- Bases Moleculares de La DM2Documento4 pagineBases Moleculares de La DM2LEONARDO ANGEL SANTOS HERNANDEZNessuna valutazione finora
- 43622-Texto Del Artículo-203119-1-10-20140522Documento10 pagine43622-Texto Del Artículo-203119-1-10-20140522keara73dNessuna valutazione finora
- Resumen Tejido Adiposo EndocrinoDocumento2 pagineResumen Tejido Adiposo Endocrinonapiyes668Nessuna valutazione finora
- Guía de Lectura - Obesidad, RI y SM - EditableDocumento30 pagineGuía de Lectura - Obesidad, RI y SM - EditableDahiana BergesNessuna valutazione finora
- Tirosina QuinasaDocumento6 pagineTirosina QuinasaSheyla Jara TorresNessuna valutazione finora
- Fisiopatología del síndrome metabólico: Resistencia a la insulina y estrés oxidativoDocumento3 pagineFisiopatología del síndrome metabólico: Resistencia a la insulina y estrés oxidativoValentina CastroNessuna valutazione finora
- Resistencia a la insulina y diabetes tipo 2Documento5 pagineResistencia a la insulina y diabetes tipo 2Gece PilcoNessuna valutazione finora
- Interacción nutriente-fármaco en diabetesDocumento57 pagineInteracción nutriente-fármaco en diabetesSHEYSHEY1993Nessuna valutazione finora
- InsulinaDocumento6 pagineInsulinaPaxZNessuna valutazione finora
- Resumen Del Artículo Bases Moleculares de Las Acciones de La InsulinaDocumento4 pagineResumen Del Artículo Bases Moleculares de Las Acciones de La InsulinaAldo AcostaNessuna valutazione finora
- Dialnet FundamentosBiomolecularesDeLaDiabetesMellitus 4788212Documento8 pagineDialnet FundamentosBiomolecularesDeLaDiabetesMellitus 4788212Rossmery RosadoNessuna valutazione finora
- BFT Reporte P6 Hipoglucemiantes Eq 2Documento8 pagineBFT Reporte P6 Hipoglucemiantes Eq 2David Valdez MondragónNessuna valutazione finora
- Las Adipocinas y Su Importancia en El EnvejecimientoDocumento5 pagineLas Adipocinas y Su Importancia en El EnvejecimientoGian Carlos Chafloque GuzmanNessuna valutazione finora
- Bases Moleculares de La ObesidadDocumento8 pagineBases Moleculares de La ObesidaddrheayNessuna valutazione finora
- UntitledDocumento4 pagineUntitledMartin GuzmánNessuna valutazione finora
- Resumen 02 BioquimicaDocumento3 pagineResumen 02 Bioquimicamiguelangel zeballos huamaniNessuna valutazione finora
- La Hiperglucemia Que Camino LlevaDocumento10 pagineLa Hiperglucemia Que Camino LlevaMatt AriasNessuna valutazione finora
- Hormonas y regulación hormonalDocumento10 pagineHormonas y regulación hormonalbryan guidosNessuna valutazione finora
- Antidibeticos Seminario 8 FarmacoDocumento43 pagineAntidibeticos Seminario 8 FarmacoMario Mayorga Duarte50% (4)
- Insulinas e Hipoglicemiantes Orales 2Documento13 pagineInsulinas e Hipoglicemiantes Orales 2Margaret RendónNessuna valutazione finora
- Cómo Actúa La Insulina, Efecto Incretina, Adiponectina y LeptinaDocumento9 pagineCómo Actúa La Insulina, Efecto Incretina, Adiponectina y LeptinaValeria VillaNessuna valutazione finora
- Diabetes Tipo IDocumento4 pagineDiabetes Tipo IAry Alvarez HurtadoNessuna valutazione finora
- Mecanismo de Accion de Insulina Con Las Tres Vias Mas EstudiadasDocumento32 pagineMecanismo de Accion de Insulina Con Las Tres Vias Mas EstudiadasVic LightYear100% (2)
- Fisiopatología Diabetes MellitusDocumento5 pagineFisiopatología Diabetes MellitusFrancisco Javier ChavezNessuna valutazione finora
- Clasificación de Dislipidemias Según Perfil Lipídico y EtiologíaDocumento33 pagineClasificación de Dislipidemias Según Perfil Lipídico y EtiologíaAlexis GuerreroGuerreroNessuna valutazione finora
- Mecanismos Que Incentivan La InsulinaDocumento17 pagineMecanismos Que Incentivan La InsulinaDIANA HELEN VERA FLORESNessuna valutazione finora
- SX MetabolicoDocumento29 pagineSX MetabolicoJeremy ChonilloNessuna valutazione finora
- Prostaglandinas y leucotrienos: mediadores lipídicos de la inflamaciónDocumento14 pagineProstaglandinas y leucotrienos: mediadores lipídicos de la inflamaciónAngela SequeraNessuna valutazione finora
- Bases Moleculares de La Diabetes Mellitus Tipo 2 La Diabetes Mellitus Es Una Enfermedad EndocriDocumento3 pagineBases Moleculares de La Diabetes Mellitus Tipo 2 La Diabetes Mellitus Es Una Enfermedad EndocrirodriguezvelaandreeNessuna valutazione finora
- Fung, L., Et Al. (2015) - Resistencia A La Insulina en La MujerDocumento12 pagineFung, L., Et Al. (2015) - Resistencia A La Insulina en La MujerTamara BravoNessuna valutazione finora
- Tejido AdiposoDocumento7 pagineTejido AdiposoJulinho M LeOnNessuna valutazione finora
- 151 495 1 PBDocumento10 pagine151 495 1 PBAimee López SantiagoNessuna valutazione finora
- Insulina y GlucagondocxDocumento22 pagineInsulina y GlucagondocxTanya Castillo ElizaldeNessuna valutazione finora
- Resistencia a la insulina y diabetes tipo 2Documento4 pagineResistencia a la insulina y diabetes tipo 2katy polancoNessuna valutazione finora
- InsulinaDocumento7 pagineInsulinaroberto.barajas9059Nessuna valutazione finora
- Sindrome MetabolicoDocumento27 pagineSindrome MetabolicoCh RoNessuna valutazione finora
- SB 3Documento15 pagineSB 3Luis Barrionuevo QuintanaNessuna valutazione finora
- Fisio InsulinaDocumento7 pagineFisio InsulinavianeyarvizuayalaNessuna valutazione finora
- Presentación Resistencia InsulinaDocumento16 paginePresentación Resistencia InsulinaNerea Trillo PérezNessuna valutazione finora
- Protocolo BQSDocumento11 pagineProtocolo BQSLily LeroNessuna valutazione finora
- Respuesta MetabolicaDocumento76 pagineRespuesta MetabolicaCarlos Taracena BaronNessuna valutazione finora
- Rompiendo La Resistencia A La Insulina - Basado En Las Enseñanzas De Frank Suarez: Estrategias EfectivasDa EverandRompiendo La Resistencia A La Insulina - Basado En Las Enseñanzas De Frank Suarez: Estrategias EfectivasNessuna valutazione finora
- Rejuvenecer Con El Plasma Sanguíneo De Los JóvenesDa EverandRejuvenecer Con El Plasma Sanguíneo De Los JóvenesValutazione: 5 su 5 stelle5/5 (1)
- Biologia Molecular de Los Transport Adores de GlucosaDocumento11 pagineBiologia Molecular de Los Transport Adores de GlucosaFabiola MastromatteoNessuna valutazione finora
- CB 70Documento1 paginaCB 70Roberto Alejandro Munoz MendozaNessuna valutazione finora
- Cabina audiométrica modularDocumento2 pagineCabina audiométrica modularRoberto Alejandro Munoz MendozaNessuna valutazione finora
- Cabina audiométrica modularDocumento2 pagineCabina audiométrica modularRoberto Alejandro Munoz MendozaNessuna valutazione finora
- Otras Bacterias EnteropatogenasDocumento92 pagineOtras Bacterias EnteropatogenasRoberto Alejandro Munoz MendozaNessuna valutazione finora
- Cabina 1Documento2 pagineCabina 1Roberto Alejandro Munoz MendozaNessuna valutazione finora
- 00 PdeC Iniciacion 2 SemDocumento22 pagine00 PdeC Iniciacion 2 SemRoberto Alejandro Munoz MendozaNessuna valutazione finora
- Cabina 3Documento3 pagineCabina 3Roberto Alejandro Munoz MendozaNessuna valutazione finora
- Unidad I El Curriculum Oculto de Las Nuevas TecnologiasDocumento17 pagineUnidad I El Curriculum Oculto de Las Nuevas Tecnologiasricardot8aNessuna valutazione finora
- Actividad de Puntos Evaluables - Escenario 2 - PRIMER BLOQUE-TEORICO - VIRTUAL - BIOLOGÍA HUMANA - (GRUPO B01)Documento8 pagineActividad de Puntos Evaluables - Escenario 2 - PRIMER BLOQUE-TEORICO - VIRTUAL - BIOLOGÍA HUMANA - (GRUPO B01)Maria Jose Arcila RuaNessuna valutazione finora
- Neurotransmisores y NeuromoduladoresDocumento21 pagineNeurotransmisores y NeuromoduladoresElizabethEsquivelCruz100% (1)
- Reporte de PracticaDocumento5 pagineReporte de PracticaShainyDiamanteNessuna valutazione finora
- Programa Respuesta Inmune 20-1Documento3 paginePrograma Respuesta Inmune 20-1Ruth Cobo RosalesNessuna valutazione finora
- Coloración Giemsa y WrightDocumento11 pagineColoración Giemsa y WrightErich Barrios C100% (1)
- Reglamento Sobre Bioseguridad, 21 de Junio de 1997Documento22 pagineReglamento Sobre Bioseguridad, 21 de Junio de 1997Edgar CalaNessuna valutazione finora
- Coccidioidomicosis: Enfermedad causada por hongos CoccidioidesDocumento16 pagineCoccidioidomicosis: Enfermedad causada por hongos CoccidioidesSegundo FernandezNessuna valutazione finora
- Evaluacion Nº2Documento4 pagineEvaluacion Nº2Pablo BascurNessuna valutazione finora
- Resumen Respectivo Del Tema 3 y 4Documento3 pagineResumen Respectivo Del Tema 3 y 4Yhamir Axel Apaza MamaniNessuna valutazione finora
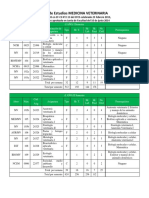
- PLAN DE ESTUDIOS FMV (Nuevo)Documento8 paginePLAN DE ESTUDIOS FMV (Nuevo)Leizi PeñaNessuna valutazione finora
- Preparación de desinfectantes para laboratorioDocumento4 paginePreparación de desinfectantes para laboratorioWendy Ortiz ArciniegasNessuna valutazione finora
- EGOyUrocultivoDocumento15 pagineEGOyUrocultivobrandon reyesNessuna valutazione finora
- GWAS y farmacogenómica: descubriendo biomarcadores para la medicina personalizadaDocumento40 pagineGWAS y farmacogenómica: descubriendo biomarcadores para la medicina personalizadaEmiliano BNessuna valutazione finora
- El Ciclo CelularDocumento12 pagineEl Ciclo CelularAlejandro FarinangoNessuna valutazione finora
- Ej09 - Respiración CelularDocumento3 pagineEj09 - Respiración CelularJosue MartinezNessuna valutazione finora
- Tejido Conectivo Mapa ConceptualDocumento1 paginaTejido Conectivo Mapa Conceptualsa11val27686% (7)
- Ugaz Saavedra Johana - Prueba Molecular 20-01Documento1 paginaUgaz Saavedra Johana - Prueba Molecular 20-01Luna U100% (1)
- Filos basales de la filogenia animalDocumento30 pagineFilos basales de la filogenia animalCamila Morillo OjedaNessuna valutazione finora
- Feria335 01 Cultivo de Tejidos Clonacion Invitro de EpidendrumDocumento20 pagineFeria335 01 Cultivo de Tejidos Clonacion Invitro de EpidendrumSamoNessuna valutazione finora
- Fisiología Respiratoria PDFDocumento7 pagineFisiología Respiratoria PDFItalo CaroNessuna valutazione finora
- Esi 2Documento4 pagineEsi 2Nerina AndrikievichNessuna valutazione finora
- CAPSICUMDocumento12 pagineCAPSICUMJESUS MARCO POZO HUAYCHANessuna valutazione finora
- Art 1Documento9 pagineArt 1José Francisco Cancino MesaNessuna valutazione finora
- Actividad de Puntos Evaluables - Escenario 5 - PRIMER BLOQUE-TEORICO - NEUROFISIOLOGIA PDFDocumento5 pagineActividad de Puntos Evaluables - Escenario 5 - PRIMER BLOQUE-TEORICO - NEUROFISIOLOGIA PDFGloriaNessuna valutazione finora
- Artículos Caroline Markolin NMGDocumento133 pagineArtículos Caroline Markolin NMGpadrino2244Nessuna valutazione finora
- Bioquímica de La Placa DentalDocumento24 pagineBioquímica de La Placa DentalLaura BustosNessuna valutazione finora
- EtimologíaDocumento5 pagineEtimologíaDiego Lopez Siquina DiegoNessuna valutazione finora
- Guía integradora UNAD de aprendizaje en BiologíaDocumento21 pagineGuía integradora UNAD de aprendizaje en Biologíayeye cardonaNessuna valutazione finora
- Actividades de CelulaDocumento2 pagineActividades de CelulaCarmela BarrazaNessuna valutazione finora
- Glosario de Terminos AmbientalesDocumento51 pagineGlosario de Terminos AmbientalesBlas HerreraNessuna valutazione finora